Cell:利用单碱基基因编辑更正人类T细胞免疫缺陷突变
CD3δ严重联合免疫缺陷(SCID)是一种危及生命的先天性免疫缺陷,是由常染色体CD3D基因的双等位基因突变引起的。CD3δ对于T细胞受体(TCR)的组装至关重要,因此,缺失CD3δ会导致严重的免疫缺陷,CD3δ SCID患者会出现严重的T细胞缺乏,常常导致婴儿期死亡。
异体造血干细胞移植(HSCT)是一种可能的治疗方法,但是可能出现致命的移植物抗宿主病(GvHD),在之前的研究中,经历异体HSCT的CD3δ SCID患者生存率仅为61.5%(n=13),并且大多数患者会出现急性GvHD,2名患者发展为慢性GvHD【1】。

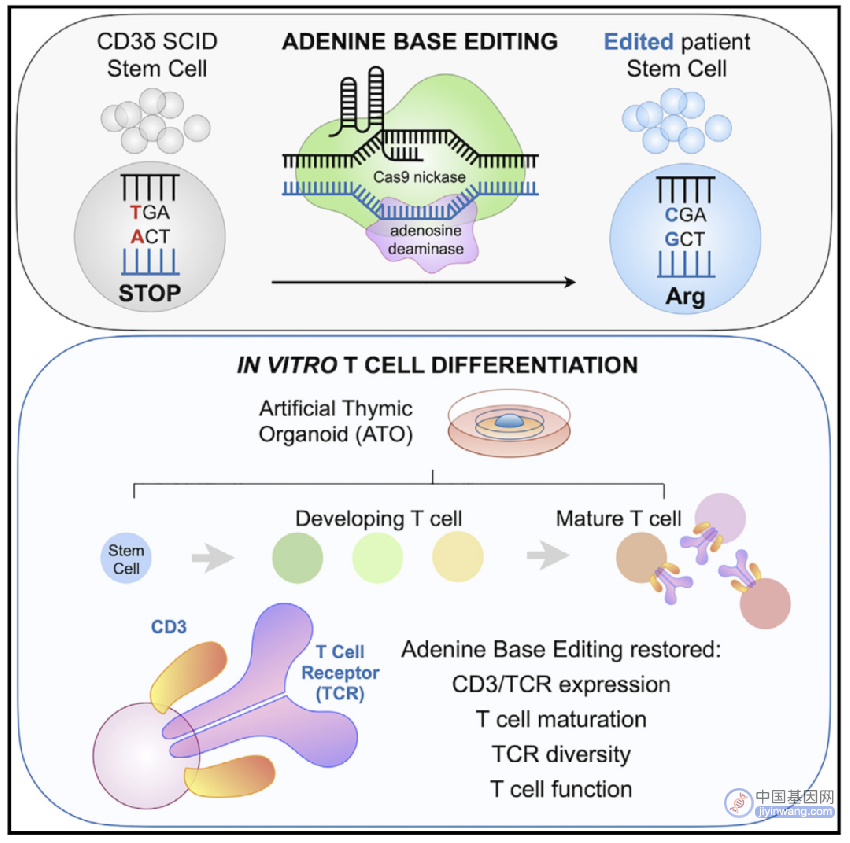
为了避免GvHD,后来人们想要利用患者自身基因改造过的造血干细胞和前体细胞(HSPC)进行自体HSCT,一开始人们通过慢病毒载体来表达基因,但是由于慢病毒会诱导致癌突变,后来人们利用CRISPR-Cas9同源修复(HDR),但目前尚未用于临床试验,而且CRISPR-Cas9需要细胞分裂来完成双链断裂和修复,难以在造血干细胞中实现高效修复,还具有脱靶的风险,而碱基编辑(BE)不需要双链DNA断裂或供体DNA模版,腺嘌呤碱基编辑器(ABE)是由催化活性受损的Cas9切口酶(Cas9n)与一个修饰DNA的脱氨酶融合在一起组成的,可以直接将A-T转变成G-C碱基对,因此可能是一个治疗CD3δ SCID的有效方法。
2023年3月20日,来自美国加州大学洛杉矶分校的Donald B. Kohn研究团队在Cell上发表题为Human T cell generation is restored in CD3δ severe combined immunodeficiency through adenine base editing 的文章,报道了ABE策略可以在自体造血干细胞和前体细胞(HSPC)中恢复CD3δ的表达,在CD3δ SCID患者的HSPC中进行碱基编辑可以更正约71.2%的致病突变,编辑后的人类HSPC移植到免疫缺陷小鼠体内可以更正约88%的致病突变,证明了ABE治疗CD3 SCID的临床前有效性。

为了研究ABE是否是治疗CD3δ SCID的合适的方法,研究人员首先构建了一个T细胞疾病模型,将CD3D突变(C202T),随后用ABE或Cas9介导的HDR进行修复,结果发现HDR的修复率为28%左右,indel副产物有大约53%,而用不同的ABE进行编辑,几乎没有indel产生,且最高的编辑率为93%。在该T细胞疾病模型中,ABE可以功能性恢复CD3δ的表达水平和信号传导。并且在CD3δ SCID患者的HSPC中用ABE进行编辑,致病突变被更正的概率大约为71.2%(n=3)。
然后他们检测了靶点附近的非目的突变和基因组上的脱靶编辑,发现ABEmax-NRTH(ABE的一个变体)是脱靶最少,最安全有效的,只有极少数的靶点附近的非目的编辑,而且并不影响健康的T细胞功能。这些编辑后的HSPC可以在人工胸腺类器官中分化,产生成熟的具有不同TCR的功能性T细胞。
接下来他们利用人类HSPC移植到小鼠模型中进行验证,首先将健康人的HSPC用慢病毒转染编辑成致病的CD3D C202T,然后电转ABEmax-NRTH进行编辑,将这些细胞移植到免疫缺陷小鼠体内,16周后从小鼠骨髓中分理出来CD34+细胞,发现有大约88%的细胞中的致病突变都被更正了,说明这些被编辑的HSPC可以重新形成造血系统,并长期维持更正致病突变的细胞群体。
CD3δ SCID的HSPC存在发育缺陷,其分化不会超过CD4 CD8双阳性的T细胞前体阶段,但是碱基编辑之后的HSPC可以挽救T细胞的发育缺陷,可以发育成为具有不同TCR的功能性T细胞。
利用ABE治疗CD3δ SCID
总的来说,这项研究证明了利用ABE治疗CD3δ SCID的临床前有效性,在体外细胞模型、患者来源的HPSC模型和人源细胞小鼠移植模型中都取得了不错的效果,且安全性也很好,期待后续的临床研究能取得成果。
原文链接:
https://doi.org/10.1016/j.cell.2023.02.027
参考文献
1. Marcus, N., et al., Hematopoietic stem cell transplantation for CD3δ deficiency. J Allergy Clin Immunol, 2011. 128(5): p. 1050-7.
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。

















 申请的区别")



