基因治疗十年一剑,诺思兰德:百亿重磅产品,解决千亿级治疗需求
1、基因治疗厚积薄发,核心产品即将进入商业化阶段
北京诺思兰德生物技术股份有限公司成立于 2004 年 6 月,并于 2021 年 11 月在北京证券交易所首批上市。

公司专注于基因治疗药物、重组蛋白质类药物和眼科用药物的研发、生产及销售。
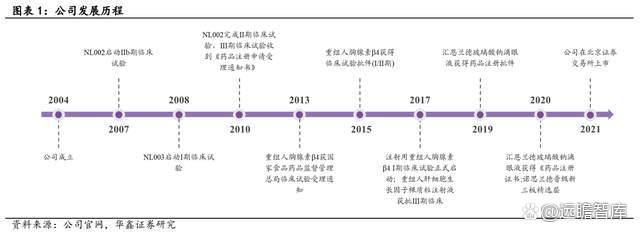
公司深耕基因治疗领域十八载,在临床前研究、临床研究、生产与质量管理、药厂建设、药品经营等方面积累了丰富的经验,并建立了基因载体构建、工程菌构建、微生物表达、哺乳动物细胞表达、生物制剂生产工艺及其规模化生产技术以及滴眼剂药物开发等核心技术平台,为后续项目的持续开发提供坚实保障。
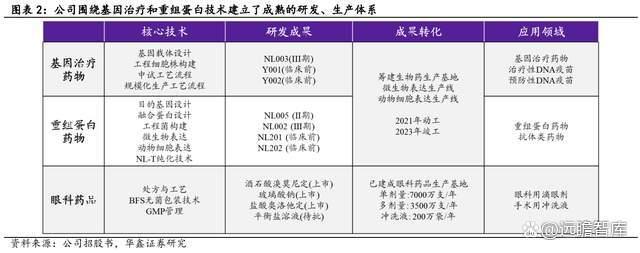
专注基因疗法与重组蛋白技术,重磅产品即将迎来兑现期。
目前,公司在研生物新药项目覆盖心血管疾病、代谢性疾病、罕见病等领域,包括 13 个生物工程新药,其中包括基因治疗药物 7 个、重组蛋白质类药物 6 个。其中,核心产品重组人肝细胞生长因子裸质粒注射液 NL003)、注射用重组人白细胞介素-11(NL002)正在进行 III 期临床试验、注射用重组人胸腺素β4(NL005)进行Ⅱ期临床试验。

以仿养创,为核心业务持续造血。
公司同时展开化学仿制药滴眼液的研制销售和滴眼液受托加工业务,由合资孙公司北京汇恩兰德制药有限公司(与韩国 Huons 合资)提供眼科产品设计、研发、生产的一体化解决方案,为新药研发提供资金支持同时积蓄开发药物市场的能力。
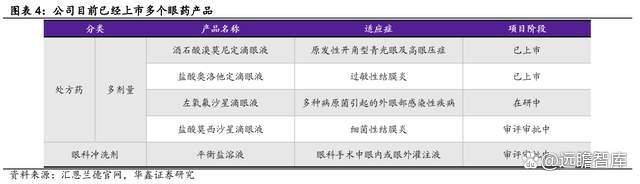
目前公司已取得 4 个滴眼液产品的注册批件,并有多个滴眼液化学仿制药项目处于研发阶段,可实现年产 1.1 亿支。
核心团队人员稳定,股权结构稳固。
截至 2022 年三季度,公司实际控制人许松山、许日山直接控股 25.57%,与其他团队创始成员聂李亚、许成日、马素永、李相哲共计控制公司股权 47.08%。公司多年以来核心人员未出现变动,持股情况稳定,核心团队稳固。

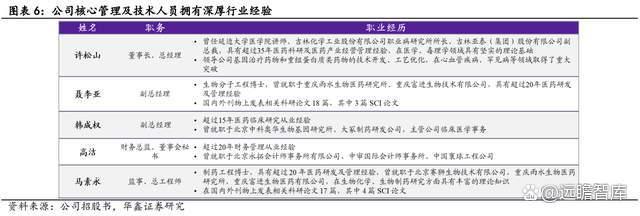
核心高层具备丰富研发和管理经验。
公司管理层和核心技术人员具备深厚的生物医药相关背景,拥有丰富研发成果,在国内外学术刊物上已累计发表文章 40 余篇(含多篇 SCI 论文)。公司董事长许松山拥有 35 年医药科研和产业管理经验,率领公司在基因治疗和重组蛋白领域取得重大突破。
公司核心技术人员聂李亚、韩成权、马素永皆拥有超过 15 年以上医药背景,全面负责产品的研究、试验、管理、审批、生产等多方面工作,是在研项目的主要驱动力。
扎实研发,创新成果累累。
公司长期以来坚持高研发投入,目前已经具备独立承担药物筛选、药学研究、临床研究与生产工艺放大等药物研发和产业化的技术体系及能力,并建立了具有领先技术水平和成本优势的生物工程新药研发和生产技术平台。
公司在裸质粒结构、工艺、质粒载体元件等部分拥有授权专利 22 项,并承担了国家级“重大新药创制” 课题 8 项。
眼药业务逐步实现盈利,为核心药物持续造血。
公司目前尚未有生物新药获批,历史上主要收入来自公司技术转让和汇恩兰德滴眼液品种的产品技术转让、生产销售。
2019 年起,公司在眼药品种上与欧康维视开展合作,后者于 2019 年 12 月和 2020 年 2 月分别获得公司玻璃酸钠滴眼液、酒石酸溴莫尼定滴眼液全国独家代理,负责该品种全国市场推广和销售;同时,2021 年汇恩兰德新产品盐酸奥洛他定滴眼液上市及盐酸奥洛他定滴眼液在第四批国家药品带量集中采购中中标,使得眼药近年的销售收入大幅提升。
目前公司仍有多个眼药品种在研,未来将不断赋能公司商业化能力,为创新药项目持续造血,保障核心业务的持续发展。
2、基因治疗大黑马,重磅产品一马当先
2.1、严重下肢缺血症危害巨大,目前尚无治愈手段
外周动脉疾病(Peripheral artery disease,PAD)中,下肢缺血性疾病在临床上最为常见,是由各种原因导致的下肢动脉狭窄或闭塞、血流灌注不足,从而导致下肢间歇性跛行、疼痛、溃疡或坏疽等缺血表现的一类疾病,主要病因包括动脉粥样硬化闭塞症(ASO)、糖尿病性动脉硬化闭塞症(DAO)和血栓闭塞性脉管炎(TAO)。

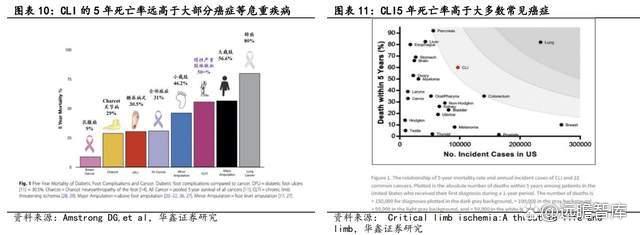
外周动脉疾病(PAD)终末期表现为重症下肢缺血(Critical limb ischemia, CLI),典型的临床表现为行走能力严重下降、静息痛(持续 2 周以上)、溃疡和坏疽,严重影响患者的生活质量,部分患者甚至只能截肢或面临死亡,其 5 年死亡率超过 50%,远高于大部分癌症等危重疾病。
我国 PAD 患者超 5000 万人,CLI 患者超 500 万人。
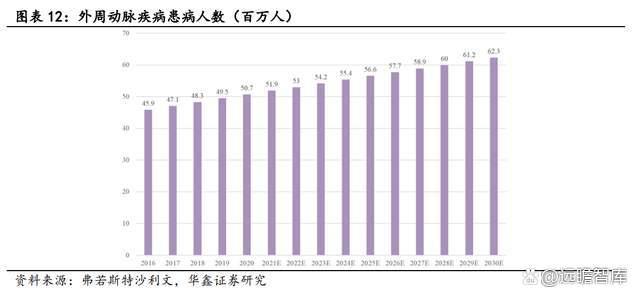
随着人口老龄化,高血压、高血脂、糖尿病及吸烟等风险因素的增长,PAD 患病风险将持续增加。根据弗若斯特沙利文数据,2020 年我国 PAD 患者人数达到 5070 万人,其中约 10-20%的患者将进入到 CLI 阶段。
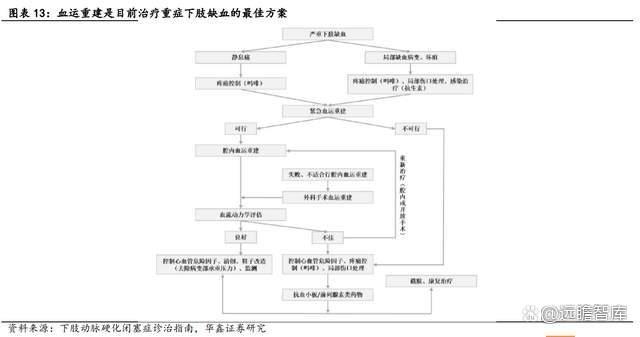
药物治疗仅能延缓病情进展,血运重建手术存在诸多限制。
传统 CLI 的治疗方法包括药物治疗、血管腔内治疗、外科手术治疗。其中,药物治疗(如抗血小板和前列腺素类药物等)仅能延缓病情进展,并不能从根本上解决动脉硬化闭塞症血管的狭窄或闭塞。目前,尚无可以彻底治愈 CLI 的药物,通过血管腔内介入或外科手术进行血运重建是治疗 CLI 的最佳方案。
根据《下肢动脉硬化闭塞症诊治指南》,当技术可行时,应对所有非截肢 CLI 患者进行血运重建。然而,血运重建亦存在诸多限制,高龄、合并疾病、下肢动脉远端流出道条件差、治疗部位再次狭窄或闭塞都可导致患者无法进行血运重建。
现有治疗手段效果不理想,临床亟需针对 CLI 新疗法的开发。
根据《外周动脉疾病管理跨大西洋学会间共识(TASCII)》数据,CLI 患者首次接受治疗中,约有 50%为外科血管重建,25%为单纯药物保守治疗,25%为直接进行截肢;而接受现有的这些治疗 1 年后的愈后情况不尽理想,CLI 治愈患者比例仅为 25%,CLI 持续进展比例为 20%,截肢比例为 30%,死亡比例为 25%。由此可见,目前 CLI 治疗手段较为局限、治疗效果差、大量医疗需求未被满足,更加有效且安全的治疗手段亟需实现突破。
2.2、基因治疗重磅单品,解决百万患者治疗需求
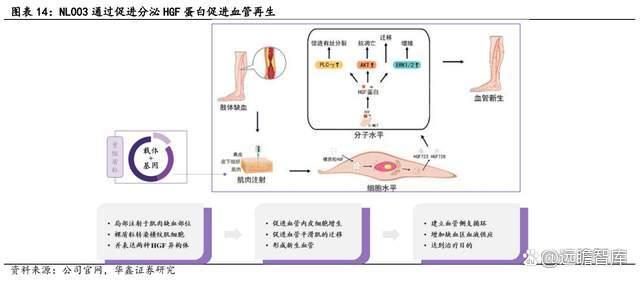
NL003 全称重组人肝细胞生长因子裸质粒注射液,主要用于治疗严重下肢缺血性疾病,包含静息痛和溃疡患者。NL003 在患者缺血部位进行局部肌肉注射,通过质粒转染横纹肌细 胞并持续表达和分泌具有促进血管生长作用的肝细胞生长因子 HGF 蛋白,促进新生血管再 生,在缺血部位形成侧支循环,建立“分子搭桥”机制,增加缺血部位的血流供应。
CLI 的传统药物治疗仅能延缓下肢动脉闭塞的病程进展,通过血管腔内介入或外科手术进行血运重建为目前最有效的治疗手段。
NL003 通过促进血管再生,可以从根本上解决动脉硬化闭塞症血管的狭窄、闭塞,从而达到治疗下肢动脉缺血疾病,因此在疗效和持久性上相对药物具有明显优势。同时与传统的血管重建手术方式比,NL003 在创伤性、操作便捷性、病人依从度、治疗费用等方面均具备优势。
NL003 的 II 期临床结果显示出优异的安全性和有效性,目前正在积极推进 III 期临床入组。
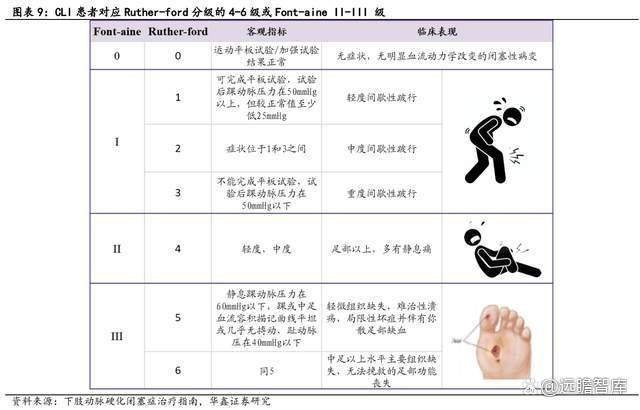
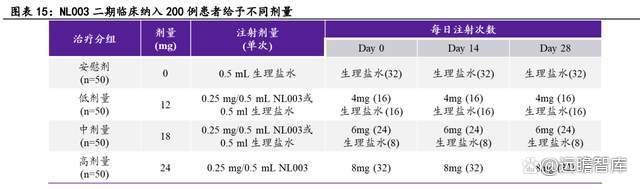
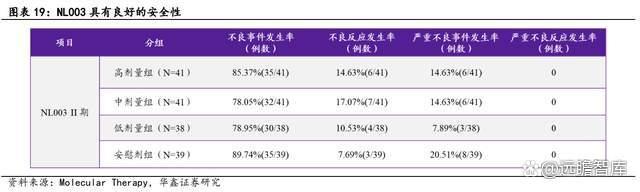
NL003 临床 II 期试验采用多中心、随机、平行、双盲、多剂量、安慰剂对照的临床试验设计,选择 Rutherford 分级为 4-5 级的 CLI 受试者,共 200 例,按 1:1:1:1 的比例,随机分成高、中、低剂量试验药组或安慰剂对照组,各组 50 例,间隔 2 周,肌肉注射给药 3 次,于 D0、D14、D28、D60、D90 和 D180 进行访视,评价患者静息痛和溃疡面积的改善情况以此评价试验药物的安全性与有效性。
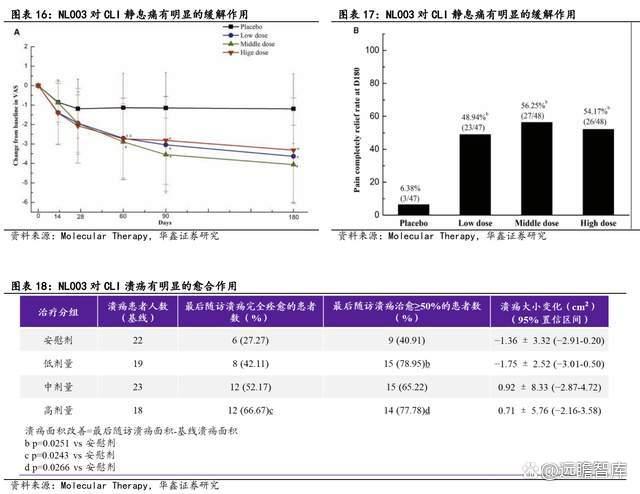
静息痛:试验结果显示与安慰剂组相比,所有 NL003 组在第一次治疗后的第 60、90 和 180 天的疼痛严重程度均有显著改善(以 VAS 测量疼痛程度)。
此外,在患者注射后第 180 天评估了停用镇痛药时疼痛完全减轻的患者比例,结果显示所有 NL003 组疼痛完全缓解的患者比例均显著高于安慰剂组(p<0.05;安慰剂组为 6.38%,高剂量、中剂量或低剂量组分别为 54.17%、56.25%或 48.94%),表明 NL003 对 CLI 静息痛有明显的缓解作用。
溃疡:低、中、高剂量组的溃疡完全愈合率均高于安慰剂组,并且随给药剂量的增加 而呈现趋势性变化,高剂量组溃疡完全愈合率与安慰剂组比较具有统计学意义 (p=0.0243),表明 NL003 对 CLI 溃疡有明显的愈合作用。
NL003 治疗 CLI 的临床 II 期试验结果显示,该药对 CLI 的疼痛和溃疡具有良好改善作用,首次给药第 60 天至 180 天,所有 NL003 给药组对比安慰剂组均能显著缓解疼痛,第 180 天后在不服用镇痛药物前提下静息痛完全消失率可达 56.25%,溃疡完全愈合率可达 66.67%,均显著高于安慰剂组。
安全性:NL003 临床 II 期试验各试验组间不良事件和不良反应发生率差异无显著差异 (p>0.05),全部不良反应分级均属于轻、中度,可自行缓解或消失,无需特殊处理;各试验组间的严重不良事件(SAE)发生率的无显著差异(p>0.05),不存在严重不良反应。
NL003 有望实现 Best in Class 潜力,具备巨大的开发价值。
➢ 优秀的有效性和安全性。
NL003 在临床 II 试验已经显示出积极的有效性结果。同时,NL003 在临床前药代动力学试验和毒理学试验,I 期和 II 期临床试验结果均显示优秀的安全性。
➢ 普及性好,奠定广泛渗透基础。
传统血运重建手术对于医生的技术要求较高,手术的质量直接影响术后效果。
NL003 采用下肢缺血部位多点肌肉注射,医护人员经过简单培训后即可完成给药,所有给药流程均可在门诊完成。同时,与传统的血管重建手术方式比,NL003 在创伤性、操作便捷性、病人依从度、治疗费用等方面均具备优势。
➢ 具备全阶段治疗潜力,潜在市场更加广阔。
PAD 是一种进展性疾病,采用药物或手术治疗的患者通常仅能保持数月到数年的时间,本身疾病仍未解决,一大部分患者随着年龄增长或不良习惯长期积累最终将发展到 CLI 阶段,面临截肢风险。
因此,如果能在早期帮助患者血管再生,恢复血液循环,理论上对于患者预后效果将有重要提升。
目前,公司已筹备间歇性行适应症扩展的研究,如若获批,则受众治疗人群将提升近 10 倍。除此之外,NL003 采用局部肌肉注射,由于 HGF 蛋白的半衰期较短,只能在给药部位局部表达 HGF,不影响其他组织细胞,安全性高,理论上亦可以用作身体其他部位,如心肌处注射,用来治疗相关疾病,潜在市场空间广阔。
➢ 结构更优,理论治疗效果更好。
NL003 可同时表达出人体内天然存在的 HGF723 和 HGF728 两种异构体,更接近于人体内的表达机制,能发挥协同作用,具有较高的生物活性。已有临床前比较研究结果表明,表达 2 种异构体的 HGF 裸质粒具有更强的生物学活性,比仅使用单一形式 HGF 更有效地诱导血管生成,理论效果更好(同类日本 Anges 与人福均表达 HGF728 一种异构体)。
➢ 临床证据更充分,支持获批后医生使用。
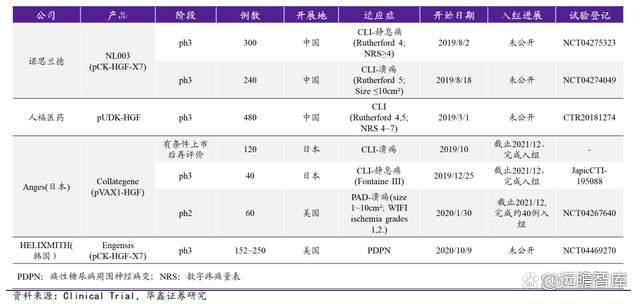
目前基于 HGF 路线研发的三家企业中,诺思兰德 NL003 的 III 期入组人数为 540 人,三者最多,远高于日本 AnGes。同时 NL003 的临床实验中纳入患者人群更广泛(包含糖尿病患者),人福 HGF 裸质粒药物在此前的临床入组中排除了糖尿病患者,而糖尿病是 CLI 重要诱因之一,该病患者相对其他患者的血管钙化更为严重、侧枝血管形成能力更差,症状与体征也相对更加严重。充分的临床证据将支持后续医生的临床使用。
➢ 适应症更广泛。
NL003 的 III 期临床适应症为静息痛和溃疡。日本 Anges 申请适应症为静息痛及溃疡的改善,但最终从厚生劳动省(MHLW)获批的适应症范围缩小为“用于在标准药物治疗不足且难以进行血运重建的慢性动脉闭塞患者的溃疡改善”。而人福 III 期适应症仅为 CLI 导致的肢体静息痛。
图表 20:多家 HGF 基因治疗药物进入临床后期阶段
2.3、重组蛋白百花盛开,具有巨大开发潜力
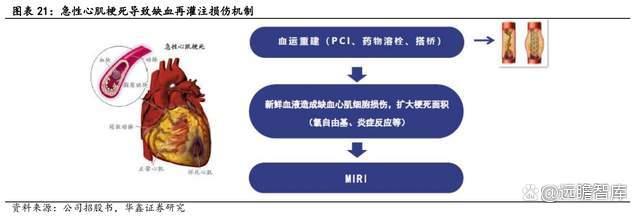
急性心肌梗死(Acute myocardial infarction,AMI)是由于冠脉急性闭塞造成的心肌坏死。目前 AMI 的主要治疗手段包括经皮冠状动脉介入治疗(PCI)、静脉溶栓治疗、冠状动脉旁路移植手术(CABG)等疗法。
许多患者经过治疗后缺血心肌得以恢复正常灌注,但其组织损伤反而呈进行性加重的病理过程,包括缺血期引起的心肌超微结构、能量代谢、心功能和电生理等一系列损伤性变化,在血管再通后表现得更加突出,甚至可发生严重的心率失常而导致猝死,这类表现称为心肌梗死所致缺血再灌注损伤(MIRI)。
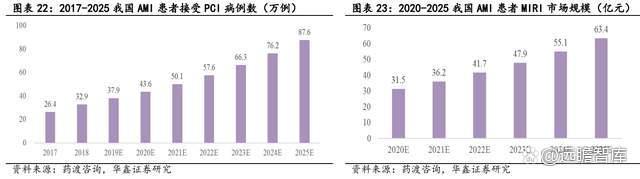
随着人口老龄化以及 PCI 手术在区县级医院的可及性的持续提升,近年来接受 PCI 手术治疗 AMI 的患者数量呈现明显上升趋势。2018 年我国有 92 万例冠心病患者接受 PCI 手术,预计 2025 年将会增长到 243 万例,其中约 36%为 AMI 患者。
根据国家心血管疾病医疗质量控制中心预测,MIRI 治疗的理论市场规模在 2025 年将达到 63.4 亿元,假设 80%患者使用该药治疗,则对应 MIRI 药物治疗市场空间可达 50.7 亿元。
全球尚无预防或治疗 MIRI 的有效药物获批上市。
若能有 MIRI 治疗药物上市,将对维护心脏功能、PCI 术后的 AMI 患者预后、提高患者生存质量具有重要意义。目前我国相关治疗药物的在研企业仅有诺思兰德和北京泰德,二者均处于临床二期阶段。

公司注射用重组人胸腺素β4(NL005)主要基于自主研发的重组人胸腺素β4 技术。研究表明 Tβ4 可通过血液循环到达心脏的再灌注损伤部位,通过调控炎症、阻止心肌细胞凋亡、缺血部位新生血管及组织修复功能,从而达到治疗 MIRI 的目的。
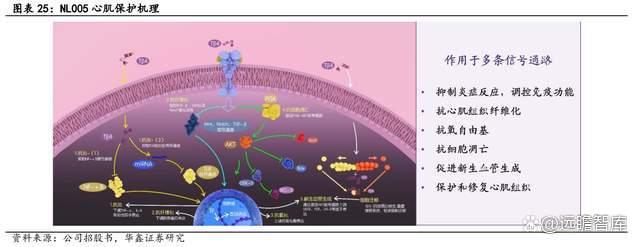
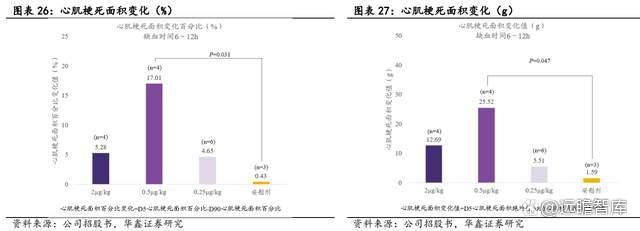
NL005 项目已经完成 IIa 期总结,积极推进 IIb 期临床试验。对缺血时间为 6-12 个小时的受试者进行的亚组分析结果表明,各给药组均可减少心肌梗死面积,其中中剂量组显著优于安慰剂组(p<0.05)。安全性方面,未发现严重不良反应,未产生抗药抗体,显示安全性良好。
改构胸腺素β4 可生产更高生物活性、更多功能性的突变体,具有多适应症开发潜力。
当前研究中针对的主要适应症为心肌梗死所致缺血再灌注损伤(MIRI)、急性肺损伤(ALI) /急性呼吸窘迫综合征(ARDS)和干眼症,分别为 NL005、NL005-2 和 NL005-1 三个项目。 研究表明,重组人胸腺素β4 还在皮肤创伤、慢性心衰中风、多发性硬化症等多个适应症 方面有开发潜力。
2.4、重组人改构白介素-11,有望成为更有效的新一代治疗品类
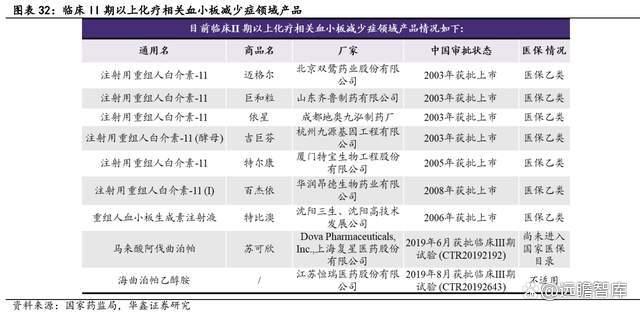
肿瘤化疗相关性血小板减少症(chemotherapyinduced thrombocytopenia,CIT)为化疗常见的不良反应,是指抗肿瘤化疗药物对骨髓巨核细胞产生抑制作用,导致的外周血中血小板计数低于一定水平(通常低于 100×109/L),发生率与肿瘤类型、治疗方案和化疗周期等有关。CIT 可能导致化疗药物剂量降低或化疗时间延迟,甚至需要输注血小板,降低化疗效果和生存质量。目前临床上 CIT 治疗的主流方法为重组人白细胞介素-11 治疗,其他方法包括血小板输注和重组人血小板生成素,但存在诸多缺点。
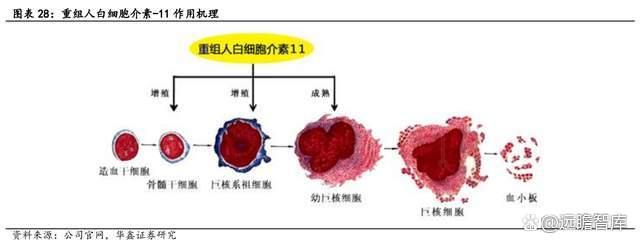
譬如,血小板输注需反复多次输注、来源短缺、血源潜在污染等缺点。而重组人血小板生成素价格昂贵且存在较多不良反应。重组人白细胞介素-11 是一种多效细胞因子,通过直接刺激造血干细胞和巨核系祖细胞的增殖,诱导巨核细胞分化成熟,促进高倍性巨核细胞生成,增加单个巨核细胞血小板的产量,促进血小板的生成,从而实现调控免疫、抗炎和保护黏膜上皮等多种功能。
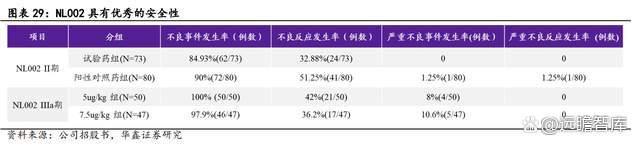
注射用重组人改构白介素-11(NL002)已经完成临床 II 及 IIIa,显示积极结果。
NL002 临床 II 期采用多中心、随机、单盲、阳性对照、自身交叉设计,共入组 88 例化疗致血小板减少患者,每名患者均接受两个周期化疗,分别使用 NL002 和对照药(已上市白介素 -11)。
试验结果表明,7.5μg/kg NL002 和 25μg/kg 对照药防治肿瘤化疗所致血小板减少症疗效相当,具有非劣效性,同时 NL002 不良反应发生率(32.88%)明显低于已上市的对照药不良反应发生率(51.25%)(p=0.0220)。试验中出现的不良反应大部分属于轻中度,可自行缓解或消失,无需特殊处理,试验过程中 NL002 组未发生严重不良事件(SAE)。
公司 NL002 的核心成分为天然结构人白细胞介素-11 的改构体,在分子结构和生产工艺上具有多项创新性,目前临床上表现出了更优的治疗潜力。
➢ 药效上具有非劣性而安全性更好。
目前上市的产品不良反应较多,如水肿、心动过速、心悸、房颤/房扑、结膜充血,且用药剂量(25-50μg/kg)较大。NL002 与已上市的多种重组人白介素-11 相比,临床有效剂量仅为同类产品的 1/3-1/5,不良反应发生率降低 40%,病人的耐受性良好。
➢ 具有改良为长效制剂的可行性。
NL002 可以使用较低剂量达到治疗效果,因此具有改良为长效制剂的可行性,可将给药方法由每日一次连续给药 7-10 天,改为每周给药一次,可以明显提高临床使用的方便性。
CIT 患者人数逐年增加,中期治疗市场规模约 50 亿。根据药渡咨询预测,我国每年接受化疗治疗的人群预计将从 2018 年的 262 万人,到 2030 年增长至 350 万人。其中,2018 年中国新发 2 级以上 CIT 患者达到 31.4 万人,预计到 2030 年中国每年新发 2 级以上 CIT 患者人群将达到 42 万人。预计 2025 年,CIT 治疗市场规模将达到 45.2 亿元。
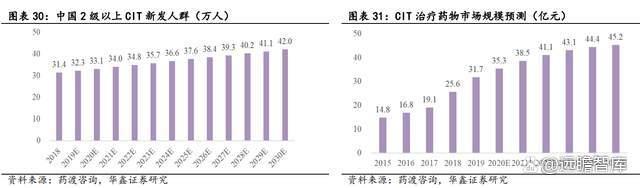
目前国内共上市 6 个厂家的注射用重组人白介素-11,在上市重组人白介素-11 中,山东齐鲁制药有限公司的巨和粒占主要市场份额,在 2018 年中国重组人白介素-11 销售额中占 62%。2019 年,我国注射用重组人白介素-11 销售总规模约为 8.5 亿元,在整个血小板减少症市场中占比为 26.8%。
3、重磅产品上市在即,即将进入产品兑现期
目前,公司核心产品 NL003、主要产品 NL005、NL002 均推进到临床中后期,其他产品尚处在临床早期阶段。其中,NL003 有望于 2023 年进行上市申请,成为公司最快实现商业化的品种。
我们预计 NL003 有望成为百亿销售的重磅大单品,未来潜在空间巨大,核心假设如下:
➢ 根据《中国心血管健康与疾病报告》数据,2021 年我国约有 4530 万下肢动脉疾病患者,参考过往 20 年我国相关患者的增长速度,预计未来每年将保持 2%的增长。
➢ PAD 患者中约有 10%-20%的患者发展到 CLI 阶段。保守假设首个预测年份 CLI 进展 比例为 10%。CLI 是非常典型的生活方式性疾病,预计随着医疗技术发展,百姓健 康意识的增强,进展率逐年减低。
➢ CLI 已经发展至下肢缺血病非常严重的阶段,患者生活质量受到严重影响,因此患者几乎都会在这个阶段寻求治疗。根据现有参考数据,75%的患者选择药物保守治疗或手术治疗,保守假设首个预测年份 70%的患者接受治疗,随着医疗技术发展,百姓健康意识的增强,接受度逐年增长。
➢ 目前,日本同类药品 Anges 的 Collategene 年治疗费用约为 8 万元/年,参考其费用,结合我国国情及现有治疗手段情况,保守设定初始定价为 5 万元/年。后续,根据目前创新药医保谈判情况进行阶梯性降价,最终价格设定为 1.3 万元/年。
➢ CLI 患者在中国拥有庞大患者人群,但缺少治愈手段,若未来适应症得以拓宽,空间有望至少翻倍,目前阶段保守估计 10 年渗透率达到 23%。
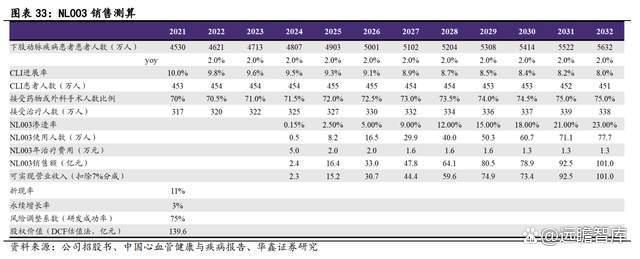
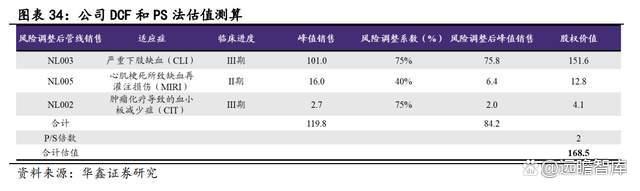
我们使用 DCF 及 PS 核心产品估值法对 NL003 的价值进行测算,通过两种方法分别估值得到 139.6 和 151.6 亿元,则保守估计 NL003 单品可支撑的合理市值为 139.6 亿元。
4、风险提示
1)研发失败风险。
公司长期发展前景很大程度上取决于现有及未来的候选药物能否成功研发。在研发过程中可能存在因研发技术路线出现偏差、关键技术难点未能攻克、研发进度缓慢等因素而导致研发失败的风险,如公司无法成功完成现有产品管线的临床开发,公司业务将受到较大影响。
2)临床试验进展不及预期。
公司临床试验推进可能受到多元因素干扰。临床试验的完成进度取决于主管部门审批、与临床试验机构等第三方的合作、临床试验中心的启动、试验所需资金筹集情况、患者招募、研究过程中方案执行、药物供应、数据处理及统计分析以及过程中与监管机构沟通等各阶段的进度,任何政策变动、临床方案调整或变更、监管机构沟通时间延长、临床试验效果不及预期或失败等,都可能对临床试验的如期完成和在研药物开发的顺利推进造成不利影响,从而对推进在研药品的开发造成不利影响。
3)市场竞争风险。
公司未来可能面临新一代产品及仿制药品的竞争。如竞争对手能够获得涉及公司候选药物的生物仿制药的上市批准,公司将面临竞争压力和潜在的不利后果。如果公司无法在创新药领域持续推出新药并保持产品的不断改进,或者无法投入更多的财务、人力资源以持续取得市场认可,即使公司的新药成功商业化,也可能随着时间的推移而变得过时从而影响公司的市场份额,进而对公司的业务、财务状况及经营业绩产生不利影响。
4)市场推广不及预期。
公司的市场营销能力尚未被市场验证,在研药品商业化存在不确定性。如公司上市销售的药品定价、定位、临床使用时机或病人选择等市场策略与临床医生或患者的实际需求存在偏离,策略制定不当或实施效果未达预期,则将对公司产品的商业化前景造成较大不利影响。
5)行业政策及政府监管风险。
公司候选药物的研究、开发、制造和商业化的所有重要方面都受到严格监管。中国国家药监局、美国 FDA、欧洲药品管理局和其他类似监管机构的监管审批程序漫长、耗时且本身具有不可预测性。如果公司不能符合严格监管标准的药品生产标准、取得监管批准在研药物商业化,公司的业务可能会受到严重负面影响。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。




















