2023年,基因疗法赛道需要思考的五大发展问题
基因治疗指通过修复致病基因,或者导入正确基因从而达到治疗效果的一种手段,这种革命性的医疗技术为治疗肿瘤、遗传性疾病、自身免疫性疾病等带来了新的希望。2022年,先后有9款基因治疗药物上市,尤其是蓝鸟生物两款基因疗法先后获FDA批准上市,提振了行业的信心,但同时其令人震撼的药品定价,也引发了行业内外热烈的讨论。
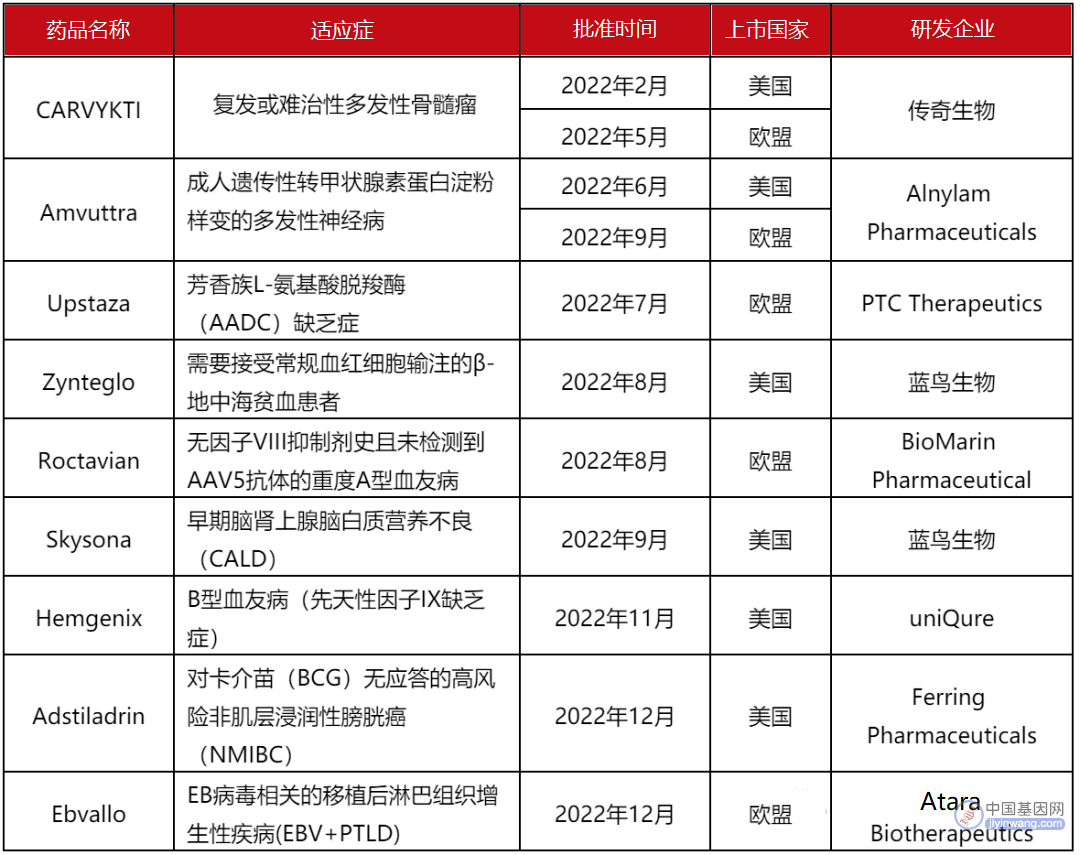
▲ 2022年基因治疗药物全球上市汇总表
Zynteglo疗法是蓝鸟生物十多年研究的成果,它的批准为蓝鸟和更多的基因治疗生物技术公司提供了验证,该领域在过去两年中一直在努力解决产品安全问题以及临床和监管挑战。蓝鸟的一位前高管这样评价道:“如果往后退一步看,我认为我们在过去几年所经历的只是成熟过程的一部分。”的确,整个基因治疗行业还非常年轻,正在走向成熟的过程中,而该领域也将在今年面临着许多下一阶段的重要问题。海外行业发展中所呈现出来的这些规律与经验,对于国内基因治疗产业的发展,将具有重要的借鉴意义。 2023年,对于全球来说,基因治疗赛道可能需要思考这么几个问题:
思考一:批准上市等于商业上的成功吗?
根据再生医学行业组织(ARM)预测,到2023年,可能有至少五种针对罕见病的基因疗法上市,包括针对镰状细胞病、杜氏肌营养不良症和血友病A的潜在新疗法。基因疗法开发商Avrobio的CEO将2023年的关键词定为“商业可行性”。这也将是蓝鸟和CSL以及今年获得批准上市的其他生物技术公司所面临的挑战。药物最终的可及性将大大影响其商业化程度,这也是细胞疗法企业正在努力突破的一个方向,对于基因疗法来说,更是如此。
首先是定价,蓝鸟和CSL都为他们的疗法设定了数百万美元的标价,理由是他们预计将为患者带来多年的益处。到目前为止,保险公司似乎并没有强烈反击,但是当基因疗法为更多患者带来影响时,情况可能会改变。到目前为止,在已上市的基因疗法中,诺华治疗脊髓性肌萎缩症(SMA)的Zolgensma可以算得上是商业化较为成功的一位,其定价为212.5万美元,去年前三季度营收10.61亿美元,同比增长10%。在美国市场,诺华采取与保险公司合作的支付策略,患者可以5年分期付款;在日本,Zolgensma被纳入健保体系,患者只需支付30%的费用;在英国,Zolgensma也被纳入了国家医疗服务体系。因此,探索创新的支付方式成为制药企业的一项重要工作。
除此之外,药企还需要证明他们能够在更大规模的治疗中提供稳定可靠的产品,这对于公司的产能规划与生产工艺等提出了新的挑战。
思考二:审评速度能否跟上管线增长速度?
以美国为例,过去数十年中大批制药公司的涌入使得未来申报临床或上市的管线会急剧增加,这将为监管机构带来大量工作。他们必须权衡临床试验的启动,并在整个开发过程中为公司提供定期的监管指导。 在今年的摩根大通医疗保健会议上,FDA生物制品评估和研究中心(CBER)宣布计划在未来两个财政年度内增加近200名新员工,并计划将专门负责基因治疗的部门——治疗产品办公室,提升为“超级办公室”地位,赋予其更多的自主权和权力。FDA的这一系列调整和举动也是向生物创新药行业发出的利好信号。
我国基因治疗产品的监管法规最早可以追溯到1993年由原国家科学技术委员会颁布的“基因工程安全管理措施”的首次发布,至今我国对基因治疗行业的监管已经经历了近30个年头,经历了最初有限制的自由发展期、全面叫停的整顿期之后,正在逐步走向鼓励发展的规范化道路。2020年12月30日,国家药典委正式出台《人用基因治疗制品总论》,该总论对基因治疗产品的生产制造、产品检定、质量控制等各环节做出详细的要求,体现出了我国对基因治疗生产要求的逐渐规范化。2021年,CDE连发两份关于基因治疗临床研究和非临床生物分布研究的技术指导原则;2022年5月发布《体内基因治疗产品药学研究与评价技术指导原则(试行)》、《体外基因修饰系统药学研究与评价技术指导原则(试行)》,监管规范化逐步加强,有效弥补了国内技术指导原则体系的缺口。
整体来说,中国的基因治疗产业还处于起步阶段,目前也暂未有产品上市,但是其发展已明显提速。截至目前,国内已获得临床试验申请的产品共有20款,包括17款AAV疗法和3款体外CRISPR基因编辑疗法。2021年,CDE仅批准了3款基因治疗产品的IND申报,2022年则增长到15款,可见基因治疗在我国的发展之快。随着我国基因治疗市场的发展增速,CDE或许也将面临FDA的“烦恼”,如何应对未来可能出现的急剧增加的临床监管需求与上市监管需求,是留给监管机构的课题。
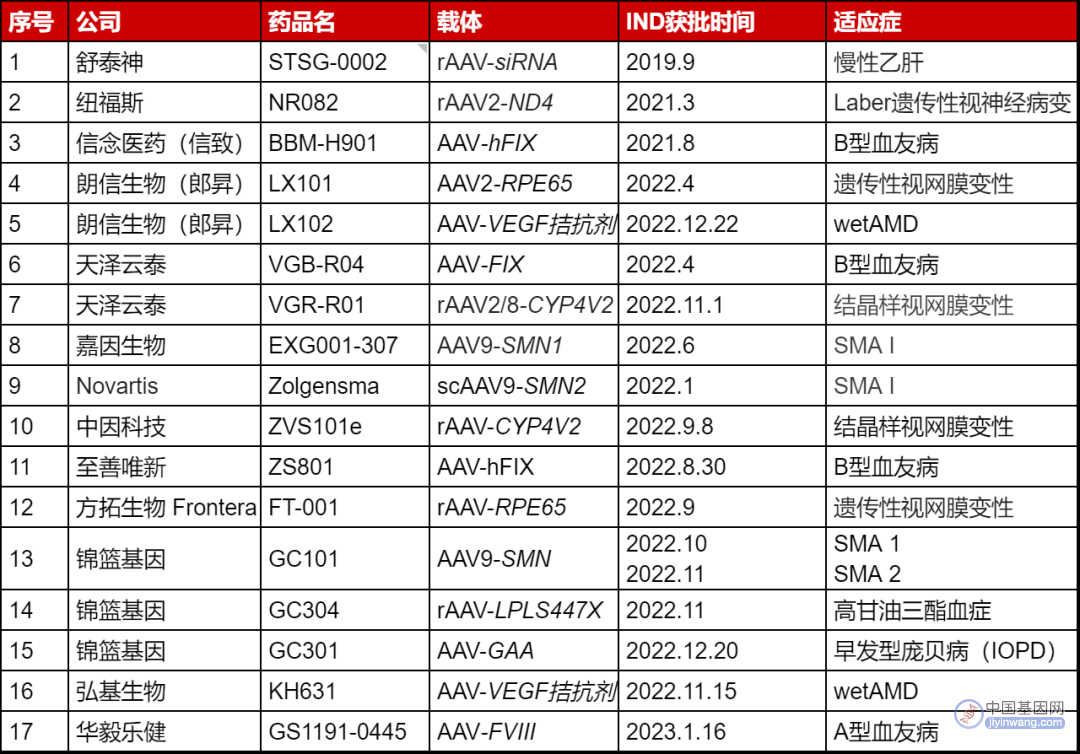
▲ 国内已获批临床的基因疗法(医麦客整理)
思考三:初创公司将如何应对长期的经济低迷?
在2022年期间,全球有十几家细胞和基因治疗公司被迫裁员,其中不乏BioMarin 和蓝鸟这样相对较大且已有临床管线的成熟公司,也有像Passage Bio和Tasyha Gene Therapies这样备受瞩目的初创公司,CRISPR基因编辑领域领先的生物技术公司Editas Medicine最近也宣布计划削减对几种临床阶段疗法的投资,并裁员20%。
基因疗法的开发成本很高,需要生物技术公司在制造方面做出昂贵的选择,并且往往越是前沿的领域,其开发成本越大。由于今年的财务前景并不明朗,该行业可能会出现更多整合,包括重组、管线重新排序、并购甚至退市或者破产。 当然,通过企业“瘦身”和战略调整,尽可能将现金跑道延长,把核心管线做到某个里程碑,也必将迎来枊暗花明。
推荐阅读:2023生物医药领域IPO会大规模重启吗?
思考四:FDA对基因编辑的看法究竟如何?
如果一切按计划进行,2023年FDA可能会首次批准CRISPR基因编辑药物。CRISPR herapeutics和Vertex Pharmaceuticals目前预计将在今年3月底前完成向该机构提交上市申请,可能会在今年晚些时候由监管机构做出相应的决定。
CRISPR和Vertex的药物是针对镰状细胞和β地中海贫血而开发的,它建立在干细胞移植的悠久历史之上,使用基因编辑技术在实验室或体外修饰患者自身的干细胞。对于十年前取得的科学突破所催化的领域而言,获得批准将代表一项了不起的成就。开发人员已经迅速采取行动,直接在体外或体内探索基因编辑。例如,Intellia Therapeutics已经报告了体内用于治疗两种罕见遗传病的CRISPR药物的临床试验结果。
但在美国,FDA对体内基因编辑仍持谨慎态度。例如,该机构搁置了Verve Therapeutics的一项研究,该研究在体内使用更新的基于CRISPR的迭代编辑工具。编辑基因时,安全性是最重要的考虑因素,这既是因为DNA脱靶的可能性,也是因为这种修饰的持久性。
Editas的首席执行官认为,监管机构理应保持谨慎,那是他们的工作。而作为药物开发企业,就是严格管理风险,确保能够推进新技术的应用。 所以,对于基因疗法中的“进阶”版块——基因编辑疗法而言,2023年将是至关重要的一年。
思考五:针对超级罕见病的药物开发该何去何从?
前正在开发的一些基因疗法针对的罕见病确实非常小众,患者群体规模不大,在一些小的国家可能仅有数千人或数百人,这意味着临床试验不仅难以招募,而且还会减少患者数量,未来的商业化就更加难以推动了。 因此,从投资回报的角度,投资者和制药企业可能均会考虑更有前景的项目。
尽管国家层面有“七年独占权”一类的鼓励政策,但是面对成百上千种不同的罕见疾病,如何解决满足这些临床需求,仍是一大问题。
蓝鸟的Skysona预计每年仅治疗10名患者,这可能也是其“天价”的一个原因,我们是否能够找到更加高效的研发方式来应对高难度开发带来的巨额成本,又或是寄希望于未来人工智能技术、机器学习技术与生物技术的深度合作带来低成本开发方式?
小结
摸着石头过河”原是一句民间歇后语,现在多被借用来表示一种科学的工作方法,表示面对新事物要本着稳妥的态度,进行探索。虽然国内基因治疗起步较晚,目前仍处于成长发展期,但由于国外的市场相对比较成熟,因此可以借鉴海外发展经验。希望通过以上的分析,可以使国内相关行业发展时可以提前布局,避免踩雷,期待国内首款基因疗法上市的那天早日来到。
参考资料:
1.https://www.biopharmadive.com/news/gene-therapy-editing-trends-outlook-2023/640234/
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





















