癌症全基因组加倍的全新机制,会绕过有丝分裂
人类健康细胞为二倍体。约30%的人类肿瘤经历过全基因组加倍(Whole Genome Doubling/Duplication, WGD)【1】,形成多倍体,这一过程是癌症基因组不稳定性的重要驱动因素。有WGD肿瘤的病人相比之下预后更差,生存期更短。然而全基因组加倍的分子及细胞机制还不清晰。
肿瘤基因组学研究表明,抑癌基因p53的变异与WGD相关,但是近半的WGD肿瘤具有野生型p53,在此类p53野生型肿瘤中E2F-Rb通路的失调与WGD成正相关【1,2】,然而背后的机理不明确。

2023年1月20日,英国弗朗西斯·克里克研究所John Diffley实验室(博士生曾京昆为第一作者)在Cell上发表文章Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication,发现原癌基因cyclin E过表达产生的复制压力可以导致有丝分裂绕过,并且抑癌基因p53对这一过程有促进作用。Cyclin E可以进一步克服有丝分裂绕过后的细胞衰老,使细胞进行完整的内源性复制而达到全基因组加倍,这一发现完善了癌症全基因组加倍的机制,为癌症的进化提供了新的思考。

Cyclin E(细胞周期蛋白E)是调控G1期到S期过渡的细胞周期蛋白。在正常生理条件下cyclin E与CDK2结合,磷酸化Rb蛋白以激活E2F转录因子来促进细胞增殖和细胞周期进展。作者们从过去的研究中发现cyclin E的过表达可以导致多倍体细胞的产生,过去这被学者推测是由于基因组部分重复复制而导致的【3】。作者们利用流式技术发现,cyclin E过表达产生的多倍体细胞其实是由于全基因组加倍(WGD)而产生的。目前已知能造成WGD的方式大概有三条:病毒感染导致的细胞融合、胞质分裂失败或有丝分裂绕过(mitotic bypass)后内源性复制(endoreduplication)。作者们利用改进的FUCCI活细胞延时成像技术对细胞周期进行可视化观测确认,cyclin E过表达的细胞经历了有丝分裂绕过(绕过有丝分裂期,从G2期直接进入G1期)而进行WGD(图1)。作者将有丝分裂绕过后的G1期命名为EC-G1期(Endoreduplication Cycle G1)。

图1. Cyclin E过表达的细胞(+ Dox)绕过有丝分裂。
Cyclin E的过表达已知会造成复制压力,这是由于其会降低DNA解旋酶MCM在染色体上的含量,导致DNA复制时复制原点的激活异常,造成DNA损伤信号的增多。作者们想知道复制压力是否和有丝分裂绕过有关,为了检验这一可能性,作者们利用了一种DNA复制酶抑制剂阿非迪霉素(Aphidicolin,Aph)来处理细胞,发现大量经过阿非迪霉素处理过的细胞进行了有丝分裂绕过,进入EC-G1期。另外通过其它原癌基因的过表达表明,自由基升高导致的复制压力也可以促进WGD。这些结果确认了不同形式的复制压力可以导致细胞绕过有丝分裂而能潜在进行WGD。
作者们观察到细胞绕过有丝分裂前经历了长时间的G2停滞,这表明G2检查点的激活。作者们用小分子抑制剂来抑制Chk1和Wee1激酶可以使G2停滞的细胞强制性进入有丝分裂,表明有丝分裂绕过依赖于G2检查点。作者们观察到有丝分裂绕过的过程中G2标记物CycB被降解,siRNA敲低实验表明E3泛素连接酶APC/CCdh1在这一过程中降解CycB。
作者们接下来探究了p53在有丝分裂绕过中起的作用。p53是抑癌基因,在维护基因组的稳定性中起重要作用,于是作者们在细胞中敲除了p53。然而令人意外的是,在复制压力下p53KO细胞几乎完全丧失了有丝分裂绕过的能力,反而在cyclin E过表达时或阿非迪霉素中进行灾难性有丝分裂(catastrophic mitosis),这些细胞的最终命运多为死亡或者产生碎裂的细胞核【图2】。通过FUCCI细胞周期监测技术测量了G2时间后发现,p53KO细胞还具有G2停滞的能力,说明p53的敲除只是消除了有丝分裂绕过的能力但并没有消除G2检查点。这些结果表明,在复制压力下p53特异性造成有丝分裂绕过。

图2. p53KO细胞在阿非迪霉素中进行灾难性有丝分裂。
作者们通过siRNA敲低实验发现CDK抑制因子p21是p53在复制压力下导致有丝分裂绕过的下游效应蛋白。p21可以抑制多个CDK。通过使用化学小分子抑制剂发现,抑制CDK1和CDK2可以恢复p53KO细胞在复制压力中绕过有丝分裂的能力。已知CDK 与E3泛素连接酶APC/CCdh1相互抑制,所以推测在复制压力下,p21抑制CDK1和CDK2后给予APC/CCdh1一个激活窗口,CycB得以在G2停滞时被APC/CCdh1降解,细胞进而绕过有丝分裂而进入EC-G1。
p53野生型的人类上皮细胞系RPE1在阿非迪霉素中会绕过有丝分裂,然而会停滞在EC-G1并且表达细胞衰老标记物。相比较之下,cyclin E过表达的RPE1绕过有丝分裂后会继续进入S期完成核内复制。作者们推测cyclin E过表达可以克服EC-G1停滞,于是在阿非迪霉素中产生的EC-G1细胞中诱导cyclin E过表达,发现这可以使停滞在EC-G1的衰老细胞进入S期并完成核内复制,在EC-G1停滞的细胞中敲低p53或p21可以达到类似的效果。
2010年洛克菲勒大学的de Lange实验组发现端粒损伤或DNA双链断裂只能在p53缺陷的细胞中引起有丝分裂绕过【4】。此次作者们发现复制压力引起的有丝分裂绕过需要p53活性,表明不同的损伤形式会通过不同的机制造成的有丝分裂绕过,端粒损伤时可能会激活更强的ATM/ATR通路来抑制CDK,从而不需要p53的活性。作者们还发现有些实验中常见的用来抑制p53的病毒致癌蛋白并不会完全抑制p53的活性,在这些病毒致癌蛋白转化的细胞中,复制压力和DNA双链断裂都可以引起有丝分裂绕过。
在本工作中,作者们以cyclin E过表达为模型,表明原癌基因过表达产生的复制压力可以引起p53依赖性有丝分裂绕过,cyclin E进一步克服EC-G1衰老,使细胞完成核内复制以达到全基因组加倍(图3)。Cyclin E为E2F-Rb通路的一环,而这一通路在大量癌症中失调,本次研究因此有助于理解癌症倍性状态进化。此外,常见化疗药物的工作机制是靠干扰DNA复制、提供复制压力,此次工作也为癌症治疗带来新思考。
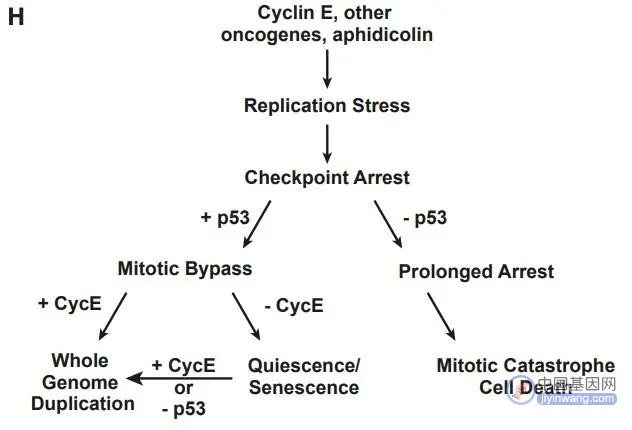
图3. 本次研究得出的模型。
原文地址:https://doi.org/10.1016/j.cell.2022.12.036
参考文献
1. Bielski, C.M. et al. (2018). Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet.50, 1189–1195.
2. Zack, T.I. et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nat. Genet.45, 1134–1140.
3. Bartkova,J. et al. (2005). DNA dam- age response as a candidate anti-cancer barrier in early human tumori- genesis. Nature 434, 864-870.
4. Davoli, T., Denchi, E.L., and de Lange, T. (2010). Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 141, 81–93.
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





")















