全球首款DMD基因疗法2月内大卖5亿,还有哪些企业能与之角逐?
杜氏肌营养不良DMD是一种罕见的遗传性肌肉疾病,在婴幼儿中发病率达1/3300,致残的几率极高。典型的杜氏肌营养不良症早在2~3岁时就会出现症状,到了12~13岁时,绝大部分患者只能依赖轮椅生活,平均寿命不足20岁。在症状表现上,它比因冰桶挑战而出圈的「渐冻症」恶性程度更高。
基因疗法攻克DMD的领军企业Sarepta与Solid Biosciences都在近日传出来喜讯,Sarepta其全球首个获批DMD基因治疗产品Elevidys在正式销售交付后的2月内大卖6910万美元(约5亿人民币);Solid Biosciences则在11月14日宣布美国FDA批准了该公司下一代DMD基因治疗候选药物SGT-003的研究性新药(IND)申请。此外,国内首家运用碱基编辑技术的基因公司新芽基因也已向FDA提交其DMD基因疗法GEN6050的PINDpre-IND申请并被受理。
全球首款上市的DMD基因疗法Elevidys大卖
今年6月22日,美国FDA批准了全球首个治疗杜氏肌营养不良症(DMD)的一次性基因疗法Elevidys,由Sarepta Therapeutics研发。Elevidys基于腺相关病毒(AAV)载体,通过基因工程办法将编码微抗肌萎缩蛋白的靶基因递送到骨骼和心肌肌肉组织,以弥补抗肌萎缩蛋白缺失。
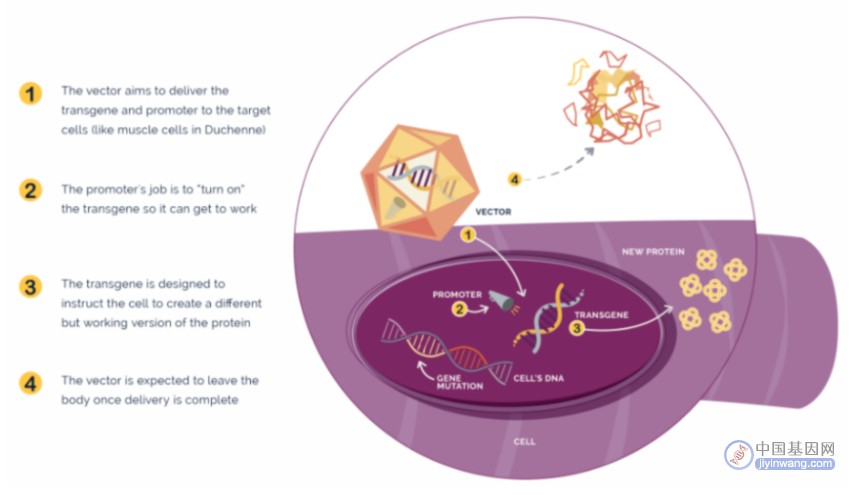
▲ 图片来源:Sarepta官网
但FDA批准的适用范围仅为4岁和5岁患儿,也导致了仅在批准Elevidys一天后,Sarepta的股价暴跌了11%。但从Sarepta 最新发布的2023年第三季度财报中,Elevidys第三季度净产品收入总计6910万美元,如果按照Elevidys在美国的定价320万美元/剂,即全球第二贵基因药物价格来推算,销售的客户约在21人左右,在8月正式销售交付的短短两个月内,这样的收益十分乐观。目前,Elevidys已经成为Sarepta公司的核心业绩增长驱动,在第三季度中的销售额已经超过该公司另外两款DMD疗法Amondys 45和Vyondys 53。
而Sarepta公司第三季度的总收入(包括净产品收入和合作收入)总计为3.318亿美元;其中净产品收入总计为3.03亿美元,同比去年增长49%。Sarepta公司称,本季度在非公认会计准则(non-GAAP)的基础上已经实现了盈利,第三季度non-GAAP净收入为3770万美元,这是Sarepta公司重要的一个里程碑。
如果能够完成Elevidys批准范围的扩大,Elevidys可能为Sarepta公司带来更大的收益,销售额有望达到40亿美元。但是在10月30日,Sarepta公司报告的Elevidys在4至7岁的 DMD 患者中展开的全球III期研究EMBARK的顶线结果,治疗后第52周的NSAA评分与基线相比的主要终点未达到,但关键次要终点达到。这对于Elevidys是否能够顺利扩展批准范围蒙上了层阴影。通常来说,如果临床研究未达到主要终点,FDA是不会认为达到次要终点的数据是积极的。在10月31日,Sarepta盘前大跌超44%。

▲ 图片来源:Sarepta官网
Sarepta 的总裁兼首席执行官 Doug Ingram 表示,“根据 EMBARK 结果,我们打算迅速采取行动,请求更新以扩大标签适应症以治疗所有患者。重要的是,我们已与 FDA 领导层分享了 EMBARK 的主要结果,他们已确认,根据全部证据,如果数据审查支持,他们对此类标签扩展持开放态度,并且他们打算在考虑后迅速推进提交的内容。”
好在,Elevidys在美国以外地区的独家权利已经被授予了罗氏,这是Sarepta与罗氏在2019年达成的协议,后续也将为Sarepta公司带来收益。
Solid的SGT-003获IND批准
2023年11月14日,Solid Biosciences宣布FDA 批准DMD基因治疗候选药物SGT-003的研究新药(IND)申请。

SGT-003是继SGT-001的第二代改进AAV基因疗法,SGT-003通过使用一种合理设计的新型亲肌AAV衣壳,称为AAV-SLB101,将Solid专有的差异化神经元型一氧化氮合酶(nNOS)微肌萎缩蛋白递送至肌肉当中,用以治疗DMD。
据披露,在小鼠和人类骨骼肌细胞体外实验中显示出优于AAV9的基因转导效率;体内试验也显示AAV-SLB101在四头肌和心肌的生物分布明显高于AAV9,同时肝脏的生物分布低于AAV9。此外,在杜氏mdx小鼠模型和体外人杜氏细胞系的研究中,观察到与AAV9相比,微肌萎缩蛋白表达增加了多倍,表明使用AAV-SLB101的治疗剂量水平可能低于第一代候选药物SGT-001。SGT-001管线已在Solid Biosciences官网上无迹可寻。
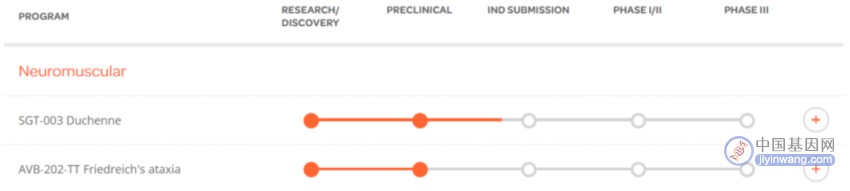
▲ 图片来源:Solid官网
基于该批准,Solid计划迅速将该研究提交给临床试验地点的美国高校的伦理审查委员会(IRB)批准,并预计在此之后不久开始患者筛查。计划中的1/2期试验SGT-003-101是首个开放标签、多中心的人体试验,旨在确定SGT-003在儿科DMD患者中的安全性和耐受性,剂量为1E14vg/kg。SGT-003将作为一次性静脉输注给两个队列的患者,每个队列至少有3名患者,并有可能扩大队列。队列1将研究4至 6岁的DMD患者,将评估治疗后5年的长期安全性和有效性。
辉瑞重金购入PF-06939926
PF-06939926是辉瑞以1.5亿美金从Bamboo Therapeutics收购的DMD基因疗法,由人肌肉特异性启动子驱动,利用rAAV9作为载体递送微型肌营养不良蛋白基因,rAAV9具有靶向定位肌肉组织的潜力。

PF-06939926目前处于3期临床阶段,在8个国家开设了14个试验点,已在西班牙、英国的试验点DMD患者已经开始给药。此前,该药已获得FDA授予的孤儿药资格和罕见儿科疾病资格。2021年9月,PF-06939926的Ib期临床试验中曾有一名年轻男性患者死亡,该试验被FDA叫停,死亡的患者接受的剂量为2×10^14 vg/kg,这提示了AAV基因治疗低剂量也可能出现严重的安全性问题。2022年4月,在辉瑞与独立外部数据监测委员会共同商议并接受该机构关于药效测定的要求并实施了方案修订后,Ⅲ期试验被批准继续进行。
国内首个DMD碱基编辑药物
新芽基因是国内首家利用碱基编辑技术进行基因治疗药物研发的公司,首个产品GEN6050旨在通过碱基编辑,达到DMD基因外显子50跳跃,恢复抗肌萎缩蛋白的表达。截至目前,全球也尚未有针对其他适应症的体内碱基编辑药物进入临床。

9月29日,新芽基因成功与TREAT-NMD药物治疗顾问委员会(TACT)完成线上咨询会议。会议围绕新芽基因的先导产品GEN6050,即将进行全球多中心的临床试验的开发计划而展开。而在2023年4月24日,新芽基因已向FDA提交GEN6050 的PINDpre-IND申请并被受理。作为最早进行体内碱基编辑药物开发公司之一,公司已经建立完善的碱基编辑药物体内外评价系统,正在针对DMD开发一系列的产品。
此外,北海康成对此适应症也有布局,其余信息暂未披露。
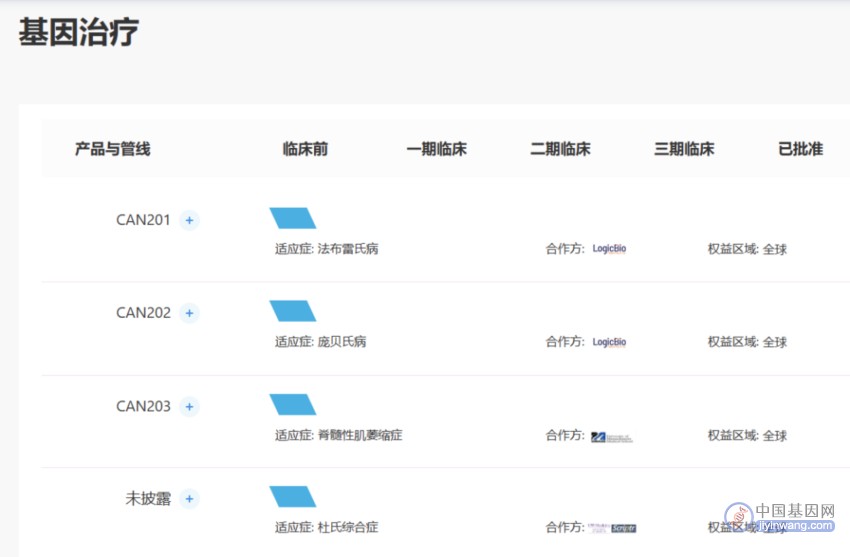
▲ 图片来源:北海康成官网
结语
DMD基因疗法赛道如火如荼展开,尽管Elevidys率先上市,千万天价仍可盈利,但有效性仍然存疑,抢跑后能否一路遥遥领先仍未可知。新芽基因另辟蹊径利用碱基编辑技术攻克DMD难题,能否早日获得IND批准走入临床,值得期待。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





















