CRISPR代谢基因筛选发现ATM激酶和KEAP1可作为肿瘤靶向治疗的靶点
近十年以来,靶向肿瘤细胞DNA损伤反应的异常或缺失在癌症治疗中取得了有效进展。其中,PARP1抑制剂在BRCA1突变患者中取得的成功加速了其他DNA修复或复制应激(replication stress)靶向抑制剂的研究【1】。目前,靶向DNA损伤反应相关的ATM激酶,DNA-PK激酶,ATR激酶等的抑制剂正处于临床试验。当发生DNA双链断裂后,ATM激酶能够通过磷酸化组蛋白H2AX等调节DNA同源重组修复和非同源连接修复。但值得注意的是, ATM激酶在代谢中也发挥着重要的作用,如氧化还原应激,戊糖磷酸途径的调节等等【2-3】。具体来讲,ATM激酶可通过磷酸化HSP27,调节戊糖磷酸途径中的关键酶葡萄糖-6-磷酸脱氢酶(G6PD)的活性,从而调节NADPH的生成【4】。另外, ATM激酶可通过磷酸化转录因子NRF1调节细胞核内线粒体基因的表达,从而调节ATP的合成【5】。因此,此前的ATM激酶抑制剂临床前研究中出现的耐药性,可能是因为癌细胞代谢途径的重编程(metabolic reprogramming)。

近日,来自NIH的Prof. Weyemi研究组在PNAS杂志发表了研究成果CRISPR metabolic screen identifies ATM and KEAP1 as targetable genetic vulnerabilities in solid tumors(博士生李浩健为第一作者)。
该研究为了揭示导致ATM抑制剂耐药性的代谢途径,采用CRISPR screen筛选三阴性乳腺癌细胞MDA-MB-231中可能造成DNA损伤响应激酶Ataxia-telangiectasia-mutated(ATM)抑制剂耐药性的代谢相关基因。筛选结果发现,Kelch-like ECH-associated protein 1(KEAP1)的缺失能够使癌细胞对ATM抑制剂更加敏感(图1a)。研究者进一步通过动物实验验证了KEAP1敲除和同时抑制ATM激酶可导致合成致死。首先,研究组采用小鼠尾部静脉注射癌细胞的方法建立了基于MDA-MB-231细胞的免疫缺陷小鼠(NOD SCID gamma)癌症转移模型,该模型可通过IVIS活体成像观察肿瘤在小鼠中的进展。当肿瘤在小鼠肺部形成后,研究者通过给小鼠口服ATM激酶抑制剂AZD0156或AZD1390的方式给药。研究者观察到相比于KEAP1野生对照组,KEAP1敲除组细胞在给药后,在肺中的进展显著降低(图1b-c)。在停止给药后,接受ATM激酶抑制剂治疗的小鼠存活率显著提高(图1d)。给药7周后的小鼠肺部组织H&E染色显示KEAP1敲除组的肿瘤在给药后显著降低(图1e)。此外,由于KEAP1在非小细胞肺癌中的突变率高达15.8%,研究者采用非小细胞肺癌细胞系NCI-H1299皮下注射建立免疫缺陷小鼠的肿瘤模型。利用该模型,研究者发现当结合Keap1敲低和口服给药时,小鼠的皮下肿瘤体积和重量显著下降(图1f)。
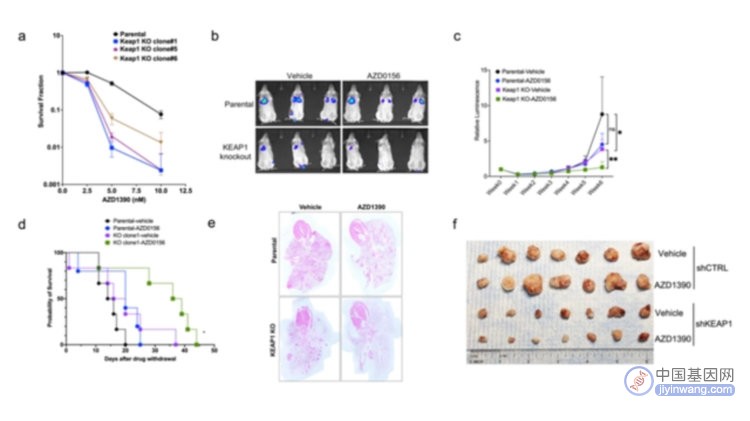
图1. 体外和体内实验验证 ATM激酶和KEAP1蛋白可作为癌症治疗的靶点。
在体外和体内实验验证了ATM和KEAP1的合成致死现象之后,背后的机制也逐渐浮出水面。KEAP1是泛素连接酶Cullin3的底物识别的适配器,其主要的底物为Nrf2蛋白,而Nrf2作为转录因子控制着超过200个与细胞氧化还原应激相关的解毒酶和抗氧化酶。当KEAP1被敲除时,Nrf2泛素化水平降低,导致下游的基因被转录激活。Nrf2通路成员之一的溶质载体家族 7 成员 11基因(SLC7A11),是胱氨酸/谷氨酸逆向转运蛋白,能够促进细胞的胱氨酸的摄取,从而增加细胞中的谷胱甘肽的合成。近年来研究表明,SLC7A11高表达癌细胞在摄取大量胱氨酸,并同时缺失NADPH后积累二硫化物应激(disulfide stress),从而导致细胞死亡【6-7】。与之相应的,研究者发现在KEAP1敲除后, SLC7A11蛋白水平得以激活,而当进一步抑制ATM激酶活性时,细胞中的NADPH含量则开始下降,从而导致反应细胞中二硫化物应激水平的GSSG含量亦显著上升以及GSH/GSSG比例下降。当研究人员用还原剂TCEP处理细胞去除二硫键时, GSH/GSSG水平和细胞存活率得到明显回升。由于TCEP处理只能部分恢复细胞的存活率,细胞内可能存在其他的致死机制。但当抑制ATM激酶时,研究人员并未发现KEAP1敲除细胞中的组蛋白H2AX磷酸化与WT细胞存在显著差异,而且抑制ATR激酶和DNA-PK激酶与KEAP1敲除亦不存在合成致死。因此,该研究中KEAP1敲除和抑制ATM激酶的合成致死的机制可能为SLC7A11激活与NADPH水平下降导致的二硫化物应激(图2),而非DNA损伤的累积或DNA修复通路的异常。
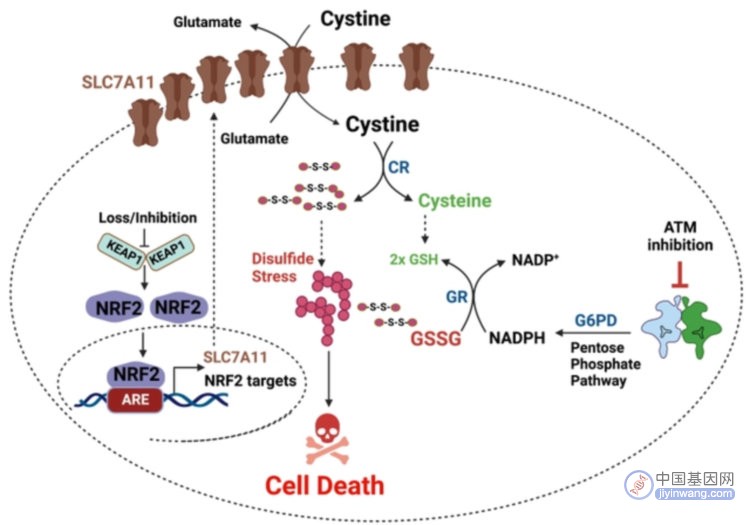
图2. 靶向ATM激酶与KEAP1蛋白的合成致死机制
综上所述
该研究发现了ATM激酶和KEAP1蛋白可作为癌症治疗的靶点,为癌症组合治疗提供了新的思路。KEAP1在所有癌症中的突变率在3.93%,尤其在非小细胞肺癌中的突变率为15.8%,本研究或为ATM激酶抑制剂在这些KEAP1突变或Nrf2通路激活的肿瘤中的应用提供依据。
原文链接:
https://doi.org/10.1073/pnas.2212072120
参考文献
1. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol.2019 Feb;16(2):81-104. doi: 10.1038/s41571-018-0114-z. PMID: 30356138; PMCID: PMC8327299.
2. Lee JH, Paull TT. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat Rev Mol Cell Biol.2021 Dec;22(12):796-814. doi: 10.1038/s41580-021-00394-2. Epub 2021 Aug 24. PMID: 34429537.
3. Li H, Zimmerman SE, Weyemi U. Genomic instability and metabolism in cancer. Int Rev Cell Mol Biol. 2021;364:241-265. doi: 10.1016/bs.ircmb.2021.05.004. Epub 2021 Jul 1. PMID: 34507785.
4. Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011 Feb 2;30(3):546-55. doi: 10.1038/emboj.2010.330. Epub 2010 Dec 14. PMID: 21157431; PMCID: PMC3034007.
5. Chow HM, Cheng A, Song X, Swerdel MR, Hart RP, Herrup K. ATM is activated by ATP depletion and modulates mitochondrial function through NRF1.J Cell Biol. 2019 Mar 4;218(3):909-928. doi: 10.1083/jcb.201806197. Epub 2019 Jan 14. PMID: 30642892; PMCID: PMC6400560.
6. Koppula P, Olszewski K, Zhang Y, Kondiparthi L, Liu X, Lei G, Das M, Fang B, Poyurovsky MV, Gan B. KEAP1 deficiency drives glucose dependency and sensitizes lung cancer cells and tumors to GLUT inhibition.iScience. 2021 May 25;24(6):102649. doi: 10.1016/j.isci.2021.102649. PMID: 34151236; PMCID: PMC8193145.
7. Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, Olszewski K, Horbath A, Chen X, Lei G, Mao C, Wu S, Zhuang L, Poyurovsky MV, James You M, Hart T, Billadeau DD, Chen J, Gan B. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol.2023 Feb 6. doi: 10.1038/s41556-023-01091-2. Epub ahead of print. PMID: 36747082.
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





















