年度医学盘点:这些“无药可治”的疾病,基因疗法带来“治愈”可能!

基因疗法通过修饰、操作或删除基因从而改变基因组的变化,有望解决传统小分子、抗体药成药性差或无法根治疾病的问题。在过去的十年间,基因疗法在癌症、遗传性疾病以及传染性疾病等多个领域的临床治疗中取得了突破性的进展。目前,针对传统方法无法治疗、预防或治愈的疾病,基因疗法让广大患者看到了“治愈”的希望。
《医学新视点》为您盘点今年以来在《柳叶刀》(THE LANCET)、《新英格兰医学杂志》(NEJM)等医学顶刊发表的与基因疗法相关的研究,以飨读者。
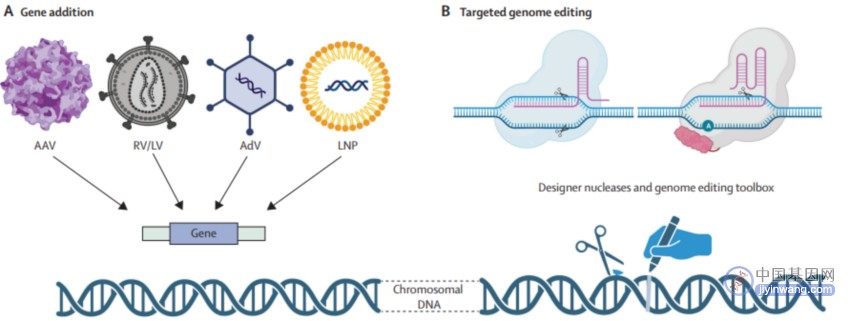
▲基因疗法整体上可以分为基因增补疗法(A)和基因编辑疗法(B)两类(图片来源:参考文献[4])
“首款”血友病B基因疗法数据公布,患者有望一针“治愈”!
以往,中度至重度血友病B患者需要通过终生、持续的凝血因子IX替代治疗来预防出血事件的发生。Etranacogene dezaparvovec是全球“首个”血友病B基因疗法,可通过AAV5载体将凝血因子IX的Padua基因变异体(FIX-Padua)递送到肝内的靶细胞,产生比正常活性高5~8倍的凝血因子IX蛋白。
一项发表于NEJM的开放标签、3期研究(HOPE-B研究)纳入了54例患有中重或重度血友病B的男性成年患者,并给予这些患者etranacogene dezaparvovec单次输注治疗。分析结果显示,患者在治疗后的几周内即可产生足够的凝血因子IX,且活性保持在较高水平。在研究导入期到治疗后的第7~18个月,患者的年出血率从4.19降到了1.51;凝血因子IX的活性在治疗后18个月随访时较基线上升了34.3%;94%的患者在接受治疗后两年内停止了所有预防性替代治疗,且患者健康相关生活质量显著改善。研究中未发现患者出现与治疗相关的严重不良反应。
值得一提的是,etranacogene dezaparvovec还有望为患者提供持久保护。另外一项发表在Blood期刊上的临床研究数据表明,单剂量基因治疗能够为中度至重度血友病患者提供超过8年的充分保护。
基因疗法面临的重要挑战之一:剂量选择
CRISPR基因编辑技术的出现和应用,为遗传性疾病的治疗带来了前所未有的希望。杜氏肌营养不良症是一种X染色体隐性遗传疾病,由X染色体上编码抗肌萎缩蛋白的Dystrophin基因突变所致。全球大约每3500名新生男婴中就有1人罹患此病,患者通常在3~5岁时开始发病(最早表现出进行性腿部肌无力),并大多在20~30岁因呼吸和心脏衰竭而死亡。
发表于NEJM的一篇论文显示,一位名叫Terry Horgan的杜氏肌营养不良症患者在接受重组腺相关病毒9型(rAAV9)载体递送的CRISPR基因编辑治疗(即CRD-TMH-001疗法)后不幸去世。该例患者在接受治疗后第6天出现急性失代偿性心衰并持续心脏骤停,并在2天后死亡。
CRD-TMH-001疗法是“首款”个体化CRISPR基因编辑疗法,也是“首款”获批临床的治疗杜氏肌营养不良症的CRISPR基因编辑疗法。研究中患者接受的静脉注射剂量为1×1014 vg/kg。尸检结果显示,患者出现了严重的急性呼吸窘迫综合征伴弥漫性肺泡损伤,肝脏中转基因的表达极少,器官中也没有腺相关病毒9型(AAV9)抗体或效应T细胞反应方面的证据。这些发现提示,患者因为对高剂量的重组腺相关病毒(rAAV)产生的先天免疫反应而死亡。在定制腺相关病毒(AAV)基因疗法时,剂量的选择仍将是一个重要挑战。
抗肌萎缩蛋白可能引起潜在免疫作用
在另外一项NEJM发表的研究中,6例杜氏肌营养不良症患者接受了重组腺相关病毒2型(rAAV2)载体递送的功能性、微型、抗肌萎缩蛋白基因疗法。分析结果发现,治疗后患者体内可检测到抗肌萎缩蛋白特异性T细胞,提供了相关基因存在表达的证据(虽然研究人员并未在患者骨骼肌中观察到功能性蛋白)。不过,在输注载体治疗前,研究人员在2例患者中意外检测到循环抗肌萎缩蛋白特异性T细胞的存在。
值得一提的是,分析还发现,回复突变体抗肌萎缩蛋白纤维(可通过缺失的内源性基因表达功能性、截短的抗肌萎缩蛋白)含有自身反应性T细胞靶向的表位。因此,未来我们在设计和监测杜氏肌营养不良症的实验性疗法时,还应当考虑T细胞对自身和非自身抗肌萎缩蛋白表位的潜在免疫作用。
《柳叶刀》重磅综述:精准基因治疗的新时代
《柳叶刀》发表的一篇综述详细总结了基因编辑技术的最新进展及其疾病治疗潜力(包括扩大治疗选择、使部分类型疾病的治疗成为可能)。该综述指出,基因疗法整体上可以分为基因增补疗法和基因编辑疗法两类。其中,基因增补疗法主要用于治疗由单一基因缺陷引起的遗传性疾病(如囊性纤维化、血友病和某些遗传性视网膜病变),该疗法通过向患者体内输送正常副本的基因,以补充缺失或不正常的基因,从而恢复其正常功能。
此外,基因编辑疗法通过编辑技术(如CRISPR/Cas9)直接在患者的DNA层面上修改或修复缺陷基因,这种方法不是添加新基因,而是更改现有的基因序列。目前,基因编辑疗法还存在很多亟待解决的问题,但该疗法在不同医疗场景中具备着广泛的应用前景。
肝脏代谢疾病中“首次”通过临床试验证明基因疗法的疗效
今年,NEJM同样公布了在研基因疗法治疗克里格勒-纳贾尔(Crigler-Najjar)综合征的1/2期临床试验结果。Crigler-Najjar综合征是一种罕见的遗传性代谢疾病,由UDP-糖基转移酶1多肽A1(UGT1A1,一种特异性肝酶)缺乏引起,导致游离胆红素在血清和身体所有组织中潜在致死性蓄积,可在大脑中产生毒性。目前光疗(phototherapy)为标准疗法,但是患者可能需要每天接受长达12小时的光疗,对生活质量产生严重影响。而且随着年龄的增长,光疗效果下降,目前“唯一”的治愈方法为肝脏移植。GNT0003利用腺相关病毒8(AAV8)载体表达编码UGT1A1的转基因,旨在通过一次性治疗,让患者的肝脏细胞自己生成UGT1A1,降低胆红素的水平。
在这项1/2期临床试验中,总计5名严重Crigler-Najjar综合征患者接受了不同剂量的GNT0003的治疗。她们在接受治疗前需要每天接受超过6小时的光疗来将血清胆红素水平控制在300 μmol/L以下。试验结果显示,接受最高剂量基因疗法治疗的3名患者的血清胆红素水平显著下降,即使在停止光疗之后,胆红素水平仍然维持在毒性水平之下,并且维持长达80周。论文指出,这是在肝脏代谢疾病中“首次”通过临床试验证明基因疗法的疗效。
3期试验数据揭示血友病A基因疗法2年结局
血友病A是由于凝血因子VIII缺乏而引起的一种罕见遗传性出血性疾病,以男性患者多见。血友病A患者通常自幼发病,有终生出血倾向,成年患者中残疾率高达80%。Valoctocogene roxaparvovec通过腺相关病毒2型(AAV5)载体,将编码凝血因子VIII基因的功能性拷贝递送到患者体内,从而帮助患者恢复自身凝血因子VIII生产能力。
此前,开放标签、单组、多中心、3期研究GENEr8-1的1年分析结果显示,134例接受高剂量(6×1013 vg/kg)的valoctocogene roxaparvovec单次给药治疗的血友病A患者,平均年化出血率下降了了84%。最新发表在NEJM的2年期结果表明,相较于接受常规预防性治疗,经valoctocogene roxaparvovec转基因治疗的重度血友病A患者2年后的年化出血率显著下降(降低幅度为84.5%)。值得关注的是,17例已接受转基因治疗长达3年的患者,仍表现出稳固疗效。
囊性纤维化患者治疗的新希望
囊性纤维化是一种常染色体隐性遗传疾病,由囊性纤维化跨膜调节因子(CFTR)基因的致病突变引起,可影响全身每个器官系统。自30多年前发现CFTR基因缺陷以来,囊性纤维化治疗的格局正在不断发生变化。THE LANCET在“2023囊性纤维化专辑(Cystic Fibrosis 2023)”中发表的一篇文章指出,近10%的囊性纤维化患者不符合使用目前CFTR调节剂药物的条件,如何满足这些患者的治疗需求是一项巨大的挑战。
目前临床科研人员正致力于通过基因替换和基因编辑策略,来满足目前不符合CFTR调节剂药物治疗条件、无法耐受现有CFTR调节剂药物的囊性纤维化患者的需求。这些治疗策略包括变异特异性核酸疗法(如针对剪接突变和无义突变的反义寡核苷酸)和变异不确定性核酸疗法(包括吸入CFTR mRNA和借助于病毒载体的CFTR基因递送疗法)。
这种“无药可治”的疾病,或将迎来“治愈”的可能!
镰状细胞病是一种常染色体隐性遗传病,由HBB基因(该基因编码成人血红蛋白[α2β2]的β球蛋白亚基)突变引起。镰状细胞病的症状通常在婴儿期出现,因为γ-球蛋白基因(HBG1和HBG2)转录转变为β-球蛋白(HBB),从而导致红细胞中胎儿血红蛋白(α2γ2)转变为成人血红蛋白。OTQ923是一种自体、离体CRISPR-Cas9编辑的CD34+造血干细胞产品,主要是通过CRISPR-Cas9系统对HBG1启动子、HBG2启动子或两者进行靶向破坏。
发表于NEJM的一项研究表明,CRISPR-Cas9破坏HBG1和HBG2基因启动子是诱导胎儿血红蛋白表达的有效治疗策略。将自体OTQ923药物输注给三例患有严重镰状细胞病的受试者,可在随访6~18个月期间持续诱导胎儿红细胞血红蛋白,并从而改善疾病的临床症状。随访结束时,所有患者均已植入并稳定诱导胎儿血红蛋白表达(胎儿血红蛋白占总血红蛋白的百分比为19.0%~26.8%)。
小 结
总体而言,基因疗法在临床上具有广泛的应用前景,其在多种“无药可治”的疾病中都展现出了优异的治疗效果,有望改变未来疾病领域治疗格局,给患者带去“治愈”新希望。
参考资料
[1] S.W. Pipe et al. Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. NEJM. Doi: 10.1056/NEJMoa2211644
[2] Angela Lek et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. NEJM. Doi: 10.1056/NEJMoa2307798
[3] Jerry R. Mendell et al. Dystrophin Immunity in Duchenne's Muscular Dystrophy. NEJM. Doi: 10.1056/NEJMoa1000228
[4] Axel Schambach et al. A new age of precision gene therapy. THE LANCET. Doi: 10.1016/S0140-6736(23)01952-9
[5] D'Antiga L et al. Gene Therapy in Patients with the Crigler-Najjar Syndrome. NEJM. Doi: 10.1056/NEJMoa2214084
[6] Johnny Mahlangu et al. Two-Year Outcomes of Valoctocogene Roxaparvovec Therapy for Hemophilia A. NEJM. Doi: 10.1056/NEJMoa2211075
[7] Simon Y Graeber et al. The future of cystic fibrosis treatment: from disease mechanisms to novel therapeutic approaches. THE LANCET. Doi: 10.1016/S0140-6736(23)01608-2
[8] Akshay Sharma et al. CRISPR-Cas9 Editing of the HBG1 and HBG2 Promoters to Treat Sickle Cell Disease. NEJM. Doi: 10.1056/NEJMoa2215643
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。




















