瞬息全碱基:为什么要研究泛基因组?
被称为泛基因组的多基因组组装可捕捉物种的遗传多样性信息,不过研究者们仍在研究构建和探索它的最佳方式。
“参考”一词流露着权威之意,提示在我们评估新信息时可将其视作可信的信源。百科全书和地图集如此,而描绘一物种染色体DNA完整序列的超高精度图谱——参考基因组(reference genome)亦是如此。
但实际上大家都知道,在呈现真实世界中的生物蓝图时,单个参考基因组往往表现不佳。西澳大学的生物信息学家David Edwards回忆起,有个同事曾想用一种经过充分研究的单一小麦品系来研究小麦遗传变异对基因表达的影响。“我们发现商品化小麦品系大约有20000个基因不存在于当时使用的那个参考基因组中。”他说。“除非考虑到这个,不然错过的可就多了。”
植物是个极端例子,它们有极其复杂多变的基因组,但隐匿的基因组多样性无处不在。有一项在64个不同人类个体基因组中开展的比较研究,揭示了近1600万个单核苷酸变异以及大于200万个由序列的缺失或插入所造成的结构变异[1]。这就使得任何一个基因组都不可能被定义为可用于比较其他基因组的“参考”。
而且,迄今为止被测序解析出的基因组大多来自西欧祖先的人,可能遗漏适用于其他遗传背景个体的基因组的关键发现。“设想某种可能的基因组学医疗实践,给某些祖先的人用疗效较好,但用于其他祖先的人时更差了,这算得上是场噩梦。”德国杜塞尔多夫大学计算基因组学研究员Tobias Marschall说,他是该比较研究的主要作者。
解决方法是泛基因组(pangenome):作为一种由多个基因组组成的复合型参考基因组,它在各染色体位点上都捕捉到更大范围的可变性和多样性信息。泛基因组此前已是用于微生物和植物研究的完备工具,现在终于要进入脊椎动物研究了。2022年7月,人类泛基因组参考联盟(Human Pangenome Reference Consortium, HPRC)发表了人类泛基因组草图的预印本,这份草图基于47位在种族和地理多样性上具有广泛代表性的人类个体[2]。还有数百个基因组现已纳入该基因组组装。
但泛基因组还太新了,这个领域仍在努力解决如何整合和探索它们等问题,以及如何劝说研究者们放下他们熟悉的传统基因组学中的线性参考。“整个领域的改变大约要花上十年。”加州大学圣克鲁斯分校计算基因组学研究员Benedict Paten预测,他也参与了HPRC。“你必须证明它确实为人们带来了些改进——不然意义何在?”
覆盖多领域
和许多遗传学领域的进展一样,泛基因组学最早也来自单细胞微生物研究。2005年,一个由美国基因组研究所的Claire Fraser和意大利Chiron疫苗公司的Rino Rappuoli共同领导的研究团队,在8株无乳链球菌(一种可导致幼儿潜在致命感染的细菌)分离株的基础上建立了基因组组装[3]。每向该组装中添加一个基因组的数据,便会向该组装中引入数十个新基因。该基因组组装被研究者们称为“泛基因组”(pan-genome,“pan”在希腊语中意为“整体”),这一名称对传统参考基因组的缺点算是直言不讳。
微生物泛基因组学是正处于蓬勃发展中的一个研究领域。美国加州大学圣地亚哥分校的系统生物学家Bernhard Palsson说,截至2013年,他的研究团队已将55种不同的大肠杆菌(Escherichia coli)菌株汇编为一个泛基因组[4]。通过评估各基因组间的变异与这些细菌的生物学功能之间存在着怎样的关联,他们能够将细菌在代谢和毒力上的差异联系到特定的基因和染色质特征。在那之后,Palsson团队将泛基因组这一概念推广到了菌株和物种水平之外,用以研究亲缘关系更远的生物,包括一类名为乳杆菌科(Lactobacillaceae)的细菌。“我们有约3500个基因组可供研究使用。”他说。
第一批真核生物的泛基因组则出现于植物学研究领域,首先是2014年由7个大豆基因组组成的基因组组装,由中国农业科学院的农作物遗传学家邱丽娟领导的研究小组创立[5]。小麦、玉米和水稻等重要农作物的泛基因组紧随其后出现。
“现在大多数主要农作物物种都有泛基因组数据了。”西澳大学的植物基因组学研究员Jacqueline Batley说,她也是Edwards的一位密切合作者。植物生物学家们正在利用这些资源开发经过改良的遗传变体,整合了与抗旱、抗病原体、增产以及其他有价值的性状相关的遗传特征。
在测序技术和基因组组装等领域的创新推动下,人类泛基因组领域已取得进展,一个由全世界研究人员组成的研究网络在2022年3月发布第一份真正完整的“端粒到端粒”的基因组序列[6]。共同领导这项工作的UCSC遗传学家Karen Miga表示,完成于2019年的首份完整人类X染色体序列(其中充斥着排列得杂乱无章的高度重复元素)就像“射向空中的一枚信号弹”,表明学界终于获得进一步探寻人类参考泛基因组的能力。她说:“这就是搞清楚如何获得正确的数据和组装策略的问题。”HPRC同年启动,Miga担任项目负责人。
进展频频
在首批泛基因组的构建中,收集DNA序列信息主要采用的是美国Illumina公司开发的“短读长(short-read)”系统。该系统虽然非常准确,但产生的读长仅有大约100-200个核苷酸。研究人员可以将这些DNA序列片段组装成“连续序列片段”(contig),从而揭示那些相对较小的差异,例如单核苷酸变体和“插入缺失”(inDel,少量核苷酸的插入或缺失),但是无法解决那些更大的结构变异。
出于这个原因,早期的泛基因组通常会将短读长测序所得的各样本contig映射到一个现有参考基因组。这种方法容易生成以基因为中心的泛基因组,缺失单个基因组中的复杂结构变异。后者可能在基因调控中发挥重要作用,而且可能蕴含了与基因组演化有关的重要信息。
尽管如此,这些“从图谱到泛基因组”的方法仍有其用处。Edwards和Batley指出,他们曾于2017年基于短读长测序分析首次尝试构建小麦泛基因组,所构建的泛基因组对于确定哪些基因不存在于或仅存在于特定品种而言颇为有效[7]。但这种也完全违背了创建泛基因组的初衷,因为该方法在选择具体以哪一个基因组作为构建泛基因组的基础时就会引入偏倚的干扰,使得同一物种泛基因组的各组装之间出现很大的不同。
更好的解决方法是先构建多个具有参考基因组质量的基因组,然后以无偏倚的方式比对这些基因组,从而描绘它们之间的位置关系以及差异所在。这一方法因而“长读长(long-read)”测序技术的快速发展而变得可行。
更长的读长也简化了另一个棘手问题。人类以及很多动植物都是二倍体(diploid),这意味着这些生物个体所携带的各条常染色体有两个拷贝。每个拷贝都有自己的变异模式,即所谓单倍型(haplotype)。有些物种的常染色体拷贝数不止两个,例如小麦就有六个。这就给短读长测序带来了一个令人困惑的问题:如何将给定的读段分配给多个染色体拷贝中的具体哪个拷贝。“这就像把两张巨大的拼图混在一起,两者的碎片都很像,你不知道某一块碎片到底属于哪张拼图。”美国洛克菲勒大学的神经遗传学研究员Erich Jarvis说。他补充说这是“获得准确的基因组数据集一个最大的问题”。
突破进行中
为了完成HPRC的泛基因组“初稿”,Jarvis和Miga及其同事借助各DNA捐赠者父母的基因组数据解决了单倍型问题,了解到各组变异的母方或父方来源[8]。长读长测序在此不可或缺,它使HPRC的科学家们得以遍历足够长的DNA片段,从而区分染色体的两个拷贝。通过将来自全部三个基因组的数据导入一个名为Hifiasm的软件工具,研究人员能够在各染色体单倍型均被明确解析(即“确定相位”)的前提下重建二倍体基因组。
尽管如此,就这一泛基因组初始版本中的47个二倍体基因组而言,它们并非像“从端粒到端粒”基因组那样可被算作完整的基因组组装。建立它们的过程中用到了一种特殊的细胞系,该细胞系所携带的两个染色体拷贝是完全相同的。而在真正的二倍体细胞中,Jarvis说,HPRC的工作流程通常产生不是一整条染色体,而是数百个大型contig,在高度相似的重复基因阵列、以及粗糙重复的着丝粒区域(该区域连接了携带基因的染色体臂)均会出现空隙。他说,HPRC仍在努力探寻处理这些问题区域的最佳方式。
好消息是现有的工作流程已实现对大部分基因组的分析,并可在很大程度上实现自动化。Marschall着重强调了一个名为Verkko的软件,是他之前的学生Mikko Rautiainen在美国国家人类基因组研究所做博士后时开发的,能够极大简化二倍体组装的过程。“有些解析出的染色体呈现出单一且定相的contig形式”,他说。这应该有助于HPRC实现其目标,到2024年时为第一代人类泛基因组组装350个基因组。
HPRC的科学家们还找到了实验方法,能帮助他们把来自同一条染色体的测序读长连接在一起(即使它们在染色体上相距甚远),消除了收集测序亲代DNA的繁琐要求。“我认为我们现在几乎可以做到用单个二倍体基因组样本就获得‘端粒到端粒’的基因组组装。”Marschall说。
这就产生了一个重要问题:如何描绘泛基因组。在过去的20年中一直被用于展示参考基因组的线性图谱,不适用于包含了数十、数百乃至数千个独立基因组的基因组组装。
该领域的大多数研究人员都把“图泛基因组(graph pangenome)”作为这个问题的最佳解决方案(见“泛基因组可视化”)。在这些精心制作的网络图中,基因组序列中的共有区域被压缩为大家熟悉的扁平直线,但在可能发生变异的位点处弯出多条不同路径。可以将其想象成一张展示了列车默认路线的城市公共交通地图。为路段养护、交通事故或运营高峰所作出的行车安排调整,可能会导致列车改道至其他线路或跳过部分车站,但在绕行的总数量上存在限制。列车线路图样式的地图可以呈现路线的不变部分,以及所有已知可能发生的绕道(本质上描绘了该线路单倍型的可能范围)。
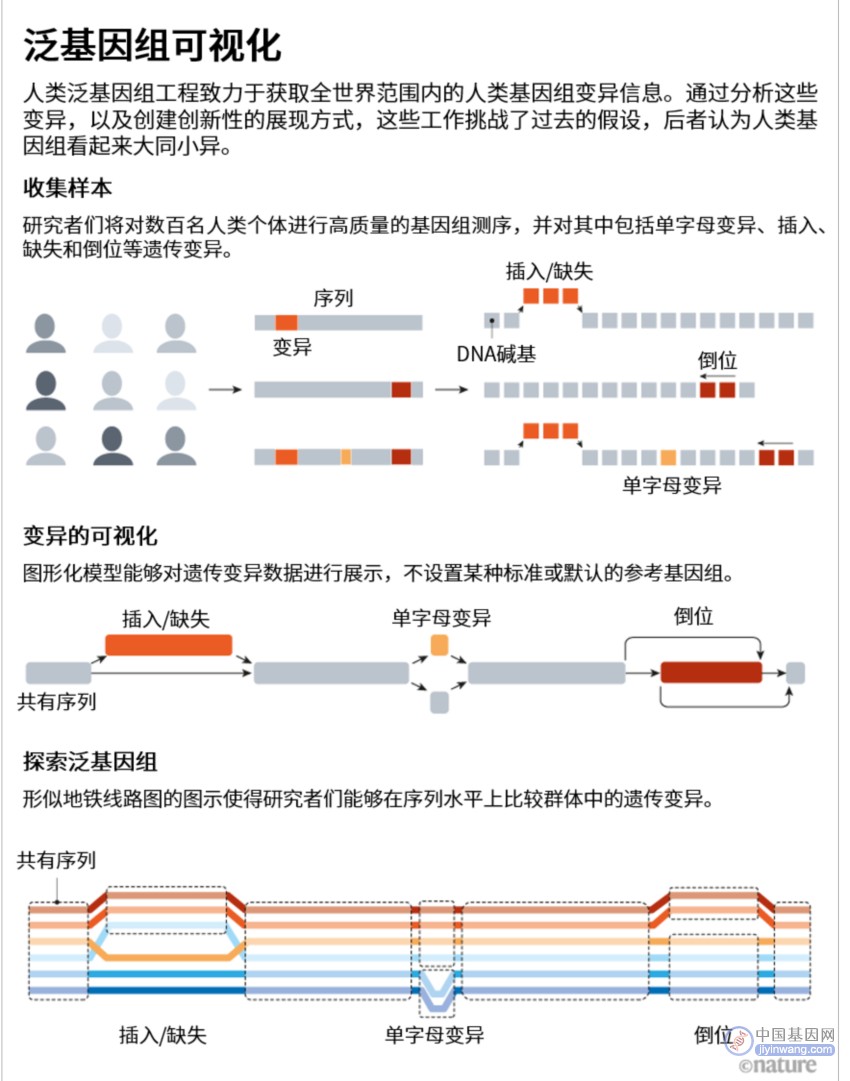
计算基因组学的研究人员仍在研究构建此类图形的最佳方法,HPRC泛基因组草图的预印本探索了几种可能的方案。其中一个方法涉及对多个独立二倍体基因组进行迭代性组装。但Miga表示,这种方法尽管可以很好地处理较大的结构变化,却“不具有精确到碱基的分辨率”。
另一种需要进行密集计算的方法是对所有基因组进行同步对齐,该方法对于包含基因的区域效果很好,但很难处理具有高重复性、低复杂性特点的那些区域。“这就是为什么这篇论文在标题中有‘草图’一词,以说明这是我们的首次尝试。”Marschall说。
而构建非人类泛基因组的研究人员们则面临着更为严峻的挑战。例如Edwards和Batley发现,围绕人类基因组而开发的图形组装软件在应用于植物时的工作效果不佳。“我们需要更多的工具”,Edwards说,指出其中一个关键问题在于植物基因组相对于人类基因组具有的更大复杂性。Jarvis目前正参与协调一项为地球上每种脊椎动物建立参考基因组的“脊椎动物基因组计划”(Vertebrate Genomes Project),他说HPRC的路径在应用至许多我们的近亲动物时效果不佳。“即使是应用于人类泛基因组,我们也发现那些组装工具需要针对不同人类个体进行一些调整。”Jarvis说。
还有一个挑战是获得来自更广泛学术社群的支持。人类参考基因组过去的几次迭代在转入日常使用上速度缓慢,许多临床实验室至今仍未采用当下最先进的一版人类参考基因组GRCh38。此外,该领域以外的研究者们可能不喜欢这种新的参考基因组格式。“人们被图形吓退了。”Batley说。
一种解决方案是构建能将图泛基因组本身藏到幕后的工具,并让研究者们能借助更为用户友好的图形界面来查询基因组的特定区域。Miga赞成在人类泛基因组与GRCh38序列坐标之间建立联系,这样当前参考基因组的使用者就无需完全修改他们的分析流程。但她补充说,促进大家接纳图泛基因组将是HPRC明年的首要任务。
全新参考架构
最终对泛基因组最好的推广,是能证明其力量的证据,而该领域的先驱者们正期待着组装良好的基因组参考所能揭开的秘密。
遥遥领先的仍是微生物泛基因组学。Palsson提及他的团队在2018年开展的一项工作,他们利用一个由近1600个结核分枝杆菌(Mycobacterium tuberculosis,结核病的致病菌)分离株组成的泛基因组对单倍型特异表型进行了分析[9]。“我们可以将它(基因组变异)与代谢特征联系起来,并阐明抗菌药物耐药机制。”他说。
与此类似的是,植物泛基因组正在帮助研究人员找到一些从前被忽视的、赋予植物恶劣环境条件下生存优势的基因。中国科学院的植物基因组学家田志喜指出,其中许多相关遗传特征存在于结构上具有可变性的基因组区域,早期的参考基因组中没有。“通常那些控制应激相关表型的基因在基因组中是重复出现的。”他说,“剂量差异会引起性状不同。”
泛基因组图谱在揭示隐匿变异上可能有同样强大的力量,这些变异隐藏于人类复杂的发育和病理状态背后。例如,临床基因组学中一般会收集数百万个微小的短读长测序片段,Paten小组的Giraffe算法可以对这些片段进行分析,并根据泛基因组图谱推断出某人的片段序列遵循哪一条单倍型“路径”,从而填补他们基因组剩下的空白部分。Jarvis还提到了有可能创建关注特定疾病和发育条件(例如自闭症谱系障碍)的泛基因组,将其与基线泛基因组进行比较,从而识别出多种不同的基因组特征。
另一个振奋人心的可能性,是将参考泛基因组与其他生物学信息相结合,从而更全面地审视染色体变异对细胞功能的影响。例如,一些研究人员正在创建“泛转录组”数据集,即用RNA测序对基因组数据进行补充,用以研究DNA变异如何影响相应基因转录产物的数量和结构。HPRC团队目前正在从其样本供体的基因组中收集表观遗传学数据,从而更好地了解基因表达在分子尺度上的个体间差异。
“这不只是碱基对的问题。”Miga强调,“我们需要着手在泛基因组基础上构建此类注释图谱,把它变成一站式的。”
参考文献:
1.Ebert, P. et al. Science 372, eabf7117 (2021).
2.Liao, W.-W. et al. Preprint at bioRxiv https://doi.org/10.1101/2022.07.09.499321 (2022).
3.Tettelin, H. et al. Proc. Natl Acad. Sci. USA 102, 13950–13955 (2005).
4.Monk, J. M. et al. Proc. Natl Acad. Sci. USA 110, 20338–20343 (2013).
5.Li, Y.-H. et al. Nature Biotechnol. 32, 1045–1052 (2014).
6.Nurk, S. et al. Science 376, 44–53 (2022).
7.Montenegro, J. D. et al. Plant J. 90, 1007–1013 (2017).
8.Jarvis, E. D. et al. Nature 611, 519–531 (2022).
9.Kavvas, E. S. et al. Nature Commun. 9, 4306 (2018).
原文以Every base everywhere all at once: pangenomics comes of age标题发表在2023年4月18日《自然》的技术特写版块上 © nature doi: 10.1038/d41586-023-01300-w
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





")















