从“最罕见变异”中找到“最易患病个体”,AI精准预测基因变异致病性
通常而言,几乎每个人都会携带有几十个潜在有害的罕见变异。无法有效识别出可能罹患疾病的高风险个体,是通过常见变异进行临床评估所遇到的最大困难之一。在灵长类基因组计划的研究中,科学家利用人工智能神经网络PrimateAI-3D,以“用最罕见的变异找到最易患病的个体”的思路,通过演化分析定位高致病性的罕见突变,并将其用于预测个体患病风险。
西班牙托马斯·马奎斯-博内特教授团队、I l l umina人工智能实验室联合多个课题组,通过对233种灵长类物种的共809个样本进行全基因组测序数据比较,鉴定出人类直系同源蛋白上430万个可能导致蛋白结构变化的基因变异位点。
研究人员以上述基因变异位点为数据集基础,将其加入人类疾病基因数据中,用包含450万种可能造成良性变异的基因数据集训练PrimateAI-3D人工智能神经网络,使之能够更准确预测基因变异的致病性。
基因变异是导致疾病的最主要原因之一。基于非人灵长类与人类的亲缘关系,相同的基因突变可能带来相似的结果,在灵长类中常见的突变可能意味着这些变异更可能是无害或危害极其有限的。
那么,如何预测一个人的遗传因素给诸如糖尿病、心血管疾病等常见疾病带来的患病风险呢?是用数千种影响较小的常见遗传变异的总和来进行评估,还是用少数影响显著的罕见突变的总和进行评估更好?
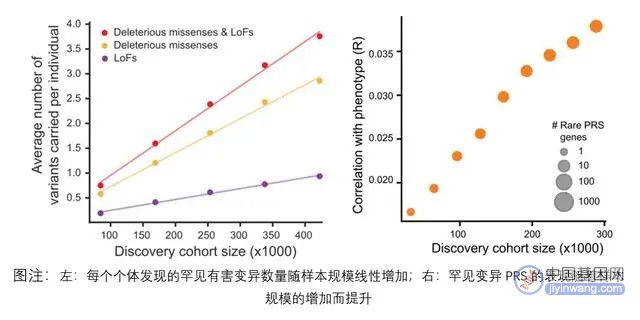
综合研究表明,常见变异和罕见变异在预测人类疾病风险方面具有互补作用。常见变异可以平均识别出更多可能患病的个体,而罕见变异更容易识别出最高风险的异常个体。因此,在临床评估上纳入罕见变异可能比仅使用常见变异更能识别出极端个体,而这些极端个体才是大部分疾病的最终病患群体,也是最需要治疗或遭受严重早期病变的群体,这对预防性筛查具有重要意义。
这一研究成功演示了将灵长类群体测序数据与深度学习模型相结合的应用,有助于我们了解人类基因变异的致病性,可帮助个性化基因组医学在临床上提供更佳诊断指导。
作者:浙江大学张国捷教授课题组集体创作、整理
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





")















