华大基因:基因科技再赋能,首个中国人血液病毒组图谱正式发布
不同地域人群和种族之间的遗传背景存在着巨大差异,因此中国人的疾病不适合用外国人的基因数据来做研究。如果把具有其他人群偏向性的知识和结论直接拿来作为中国人的疾病风险评估、遗传咨询或诊断治疗依据,不具有完善和可靠性。因此,中国代谢解析计划ChinaMAP (China Metabolic Analytics Project)应运而生,ChinaMAP一期研究覆盖全国27个省份和直辖市,8个民族,对超过1万人的高深度(40x)全基因组测序数据和表型进行系统分析。
日前,《Cell Discovery》发表了题为“The blood virome of 10,585 individuals from the ChinaMAP”的文章,该研究利用ChinaMAP数据,首次全面对中国人血液病毒组进行解析。

一、研究背景
了解人类血液病毒组对安全输血和传染病监测和控制至关重要。利用测序技术对自然种群进行调研,能够检测已知和新的病毒病原体,可为流行病学和病毒感染预防、疫苗开发和病毒基因组研究提供重要信息。此外,包括EB病毒、乙型和丙型肝炎病毒(HBV和HCV)以及人乳头瘤病毒(HPV)等在内的病毒是许多常见癌症的诱发因素。群体病毒组的地理和遗传多样性可用于公共卫生筛查和预防 。
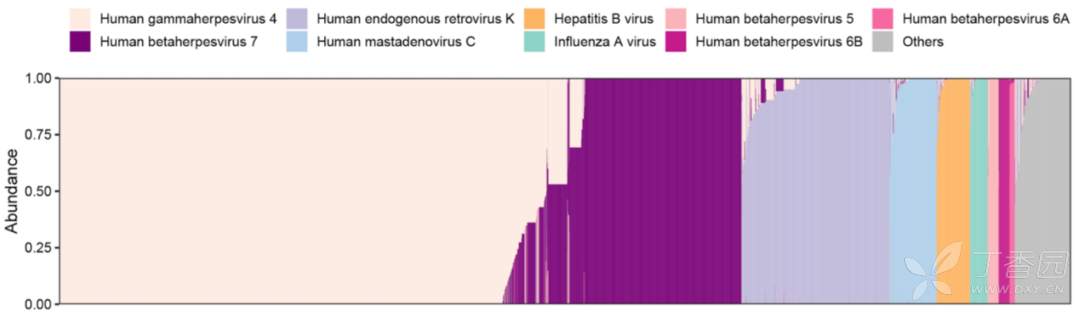
图1 病毒在血液中的分布非常丰富
二、研究方法
研究以中国代谢解析计划(ChinaMAP)的10585人数据为研究对象,对全基因组测序数据集中非人源性reads进行深入挖掘,用以调查中国人群的血液病毒组。
三、研究结果
1.血液中病毒的流行率及低于分布
在ChinaMAP数据集的 unmapped reads中 发现了14种病毒,其病毒基因组覆盖率为> 10% 。在76.7%的个体中发现了最流行的细环病毒,包括TTV(输血传播病毒)和TTV样迷你病毒(TLMV)。图3显示了血液中13种病毒的流行率、reads丰度和基因组覆盖度。在30.3%的中国人中检测到人γ疱疹病毒4 (EBV),是欧洲队列的两倍多。
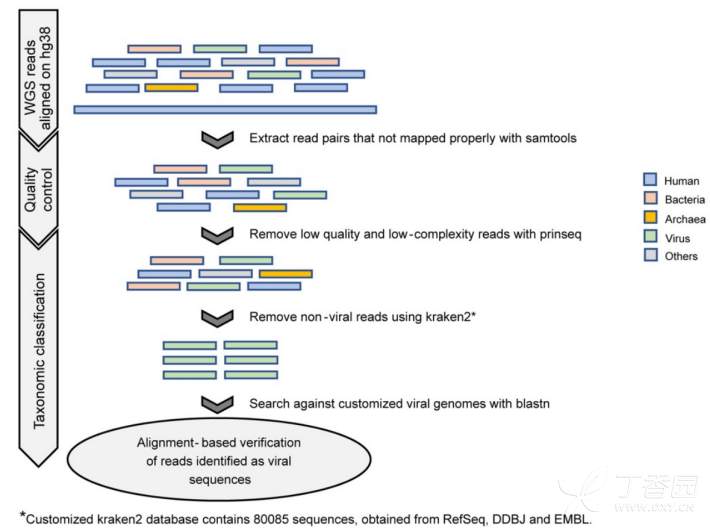
图2 从 unmapped reads中识别病毒序列的分析流程
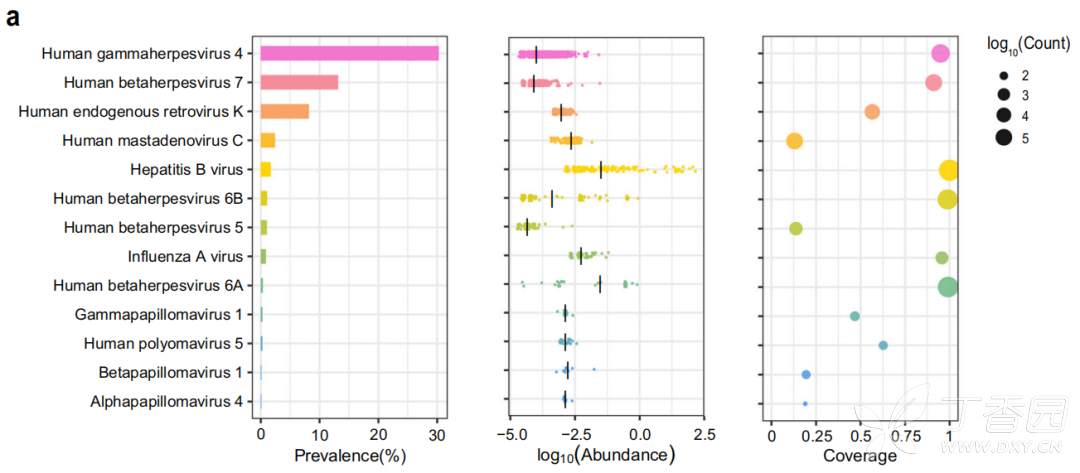
图3 人类血液中13种病毒的流行率、丰度和基因组覆盖率
人疱疹病毒7 (HHV7)、HHV6A、HHV6B和HHV5 (HCMV)分别在13.2%、0.36%、1.09%和1.03%的个体中检出。人内源性逆转录病毒K (HERV-K)、人乳突腺病毒C和HBV分别在8.20%、2.41%和1.69%的个体中检出。HPV亚型γ乳头瘤病毒1型、β乳头瘤病毒1型和α乳头瘤病毒4型在50例个体中被发现。值得注意的是,在29个个体的梅克尔细胞癌中发现的致癌性人类多瘤病毒5。在93个个体中检测到甲型流感病毒的DNA序列,这可能是由于注射了重组流感疫苗或内源性逆转录病毒组分造成的。此外,在结果中发现了非人源性病毒,经过排查发现是来自于环境和实验室的污染,因此将这部分结果归结为假阳性。
各省的人均病毒数没有差异。研究者分析了感染者中流行的前6种病毒的病毒丰度,发现HHV7和EBV在40-60岁人群中没有性别差异,研究者还分析了地理分布及相应的感染病毒数量和百分比。此外,在血液病毒组中,共检测到来自4目12科88属179个噬菌体,包括正常肠道病毒噬菌体CrAss噬菌体和大量葡萄球菌噬菌体。
2. HBV的流行率
179例HBV感染患者主要为HBV- b亚型(79例)和HBV- c亚型(59例), HBV-B在中国南方(广东、广西、福建)较为普遍,而HBV-C主要分布在西藏 (图4)。研究者分析了人类基因组中的病毒整合事件,发现10例HBV-B阳性整合,18例HBV-C阳性整合。整合事件与较高的病毒丰度显著相关,图5展示了检测到的整合断点在人类和HBV基因组中的分布。发现HBV整合位点随机分布,在人类基因组中没有明显的整合热点。然而,HBV基因组的整合断点显示在HBV gp3基因的末端和核心基因的开始处达到峰值。
构建 56 个个体 ( 每个样本变异数 ≥10 ,等位基因频率 ≥1 0%) 的对应地理来源的 HBV 序列和 20 个参考株的系统进化树 (图 6 ) 。 观察到 HBV-B 和 HBV-C 两个不同的聚类,这与 20 个参考菌株的基因型一致。 研究者 还确定了对 HBV 相关肝癌发生至关重要的 HBV 突变,包括基础核心启动子 (BCP) 突变 T1762/A1764 和核前区 A1896 突变。 HBV-C 的 BCP 突变率 (15.3%) 高于 HBV-B(2.5%) ,这与以往的报道一致 。
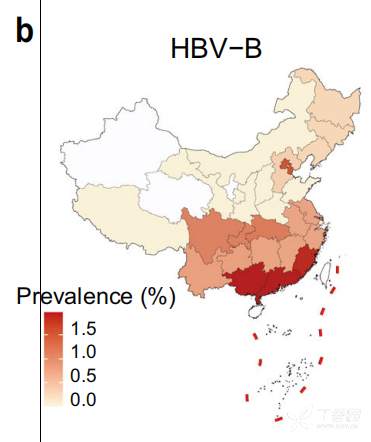
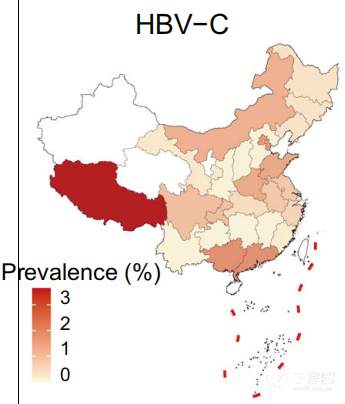
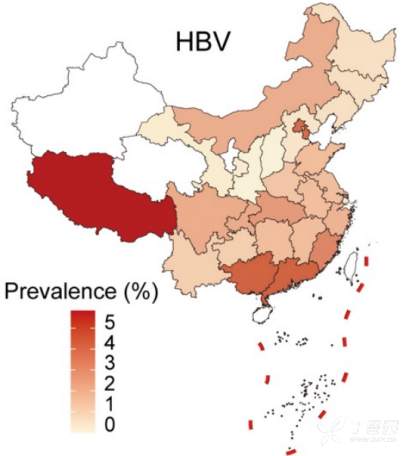
图4 HBV-B、HBV-C、HBV在中国的地理分布
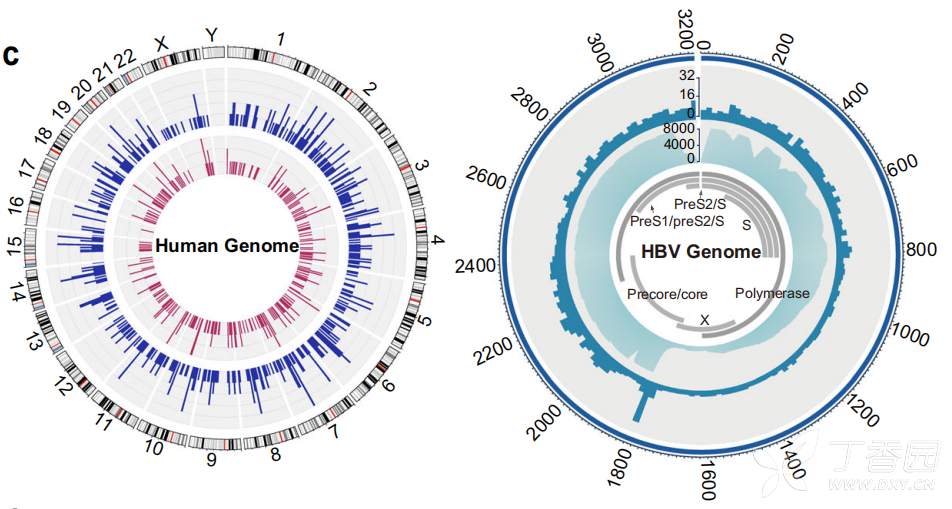
图5 乙肝病毒在人体内的整合位点(左)和乙肝病毒基因组(右)
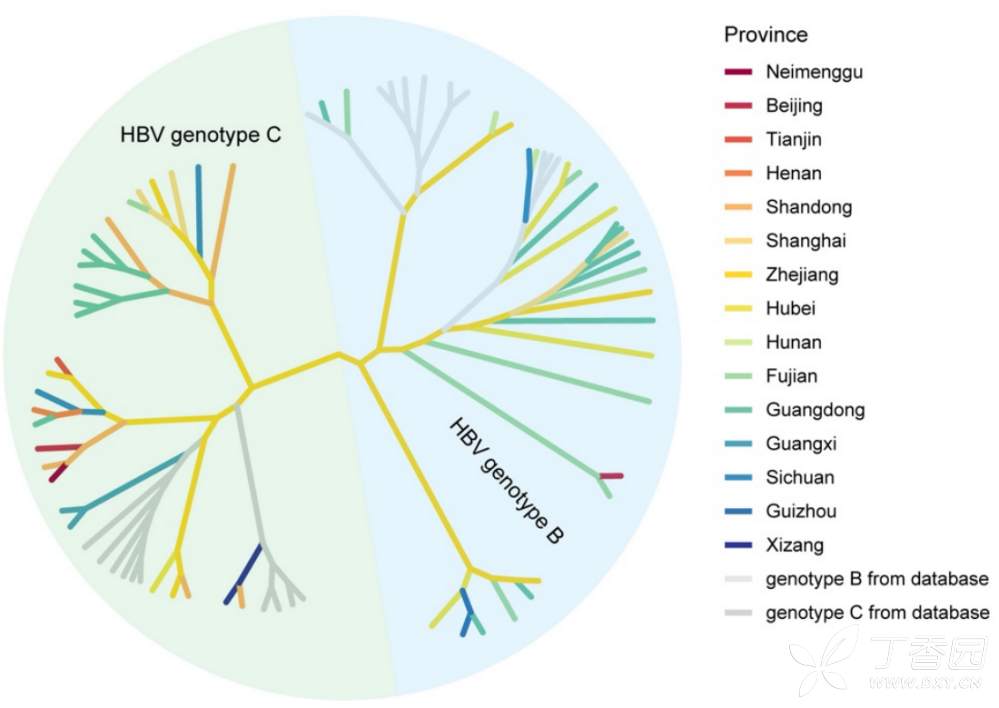
图6 HBV突变的系统发育树
3. HBV的整合事件与血液中病毒丰度相关
地 理分布显示人类疱疹病毒 HHV4 (EBV) , HHV5, HHV6A/B 和 HHV7 。 HHV6A/B 在 河北、陕西、山西和河南的发病率较高。 研究者 进行了全基因组关联研究 (GWAS) ,以探索病毒感染的遗传易感性变异。 值得注意的是,ACR 基因中的一个错义变体与 HHV6 感染显著相关 ( 图 7) 。
先前的一项研究使用 141431 名中国妇女的 低深度测 序数据 (~0.1×) 报告了感染人群中 HHV6 染色体整合与 22q13.33 靠近ACR 基因区域的 MLC1-MOV10L1 位点之间的强烈关联。ACR 编码的顶体蛋白能够促进精子穿透透明带,错义突变位于顶体蛋白 轻链中紧挨着二硫键位点 (C25) 和糖基化位点 (N22) 的高度保守区域, 表明它可能影响顶体蛋白的功能 ( 图 8) 。 HHV6A/B 可以选择性地与精子顶体完整的精子头结合,并通过性接触传播到子宫。 在 198 名丹麦捐献者的精子中,有 13.5% 的精子中存在最常见的 HHV6A/B 型疱疹 病毒。 推测ACR rs79314756 可能通 过与膜辅因子蛋白和卵母细胞受体结合,增强 顶体蛋白 活性,从而促进 C3 蛋白的切割,促进 HHV6 病毒的传播 。
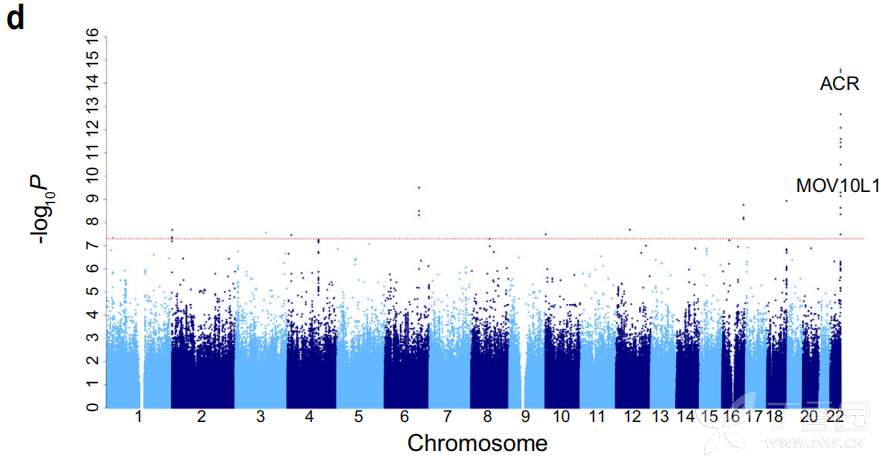
图7 HHV6感染的GWAS曼哈顿图
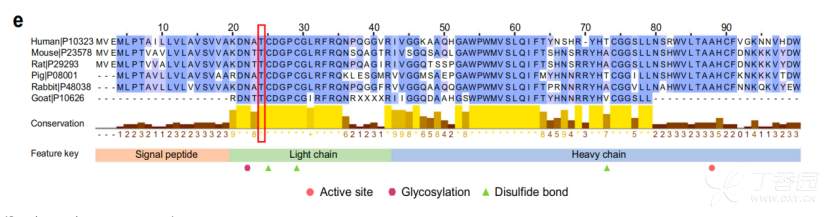
图8 顶体蛋白结构域的保守性分析
四、研究结论
总体而言,研究利用高深度的WGS数据阐明了中国人群队列中人类血液病毒组特征。分析了14种病毒的流行率、病毒丰度和地理分布。结果显示,在30%的个体中,EBV是最常见的致病性病毒,HBV的整合事件与血液中病毒丰度相关。研究发现了一种新的ACR变异与HHV6感染显著相关,为了解HHV6传播的易感性和发病机制提供了新的见解。这些发现可为预防传染病和公共卫生输血安全提供重要信息。
参考文献 :
The blood virome of 10,585 individuals from the ChinaMAP.Cell Discovery,2022.
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





















