为海量基因数据一体化分析提供新思路,哈佛开发罕见变异检验工具
“罕见变异检验方法 STAAR 发表后,受到了广泛关注。身边的许多合作者纷纷发邮件,问我们该如何使用 STAAR 这样的分析工具。
这让我们意识到,技术诞生和通过技术批量产出科技成果之间其实存在距离,就如同电动车制造技术和第一条特斯拉生产线诞生之间存在数十年差距一样。因此,我们决定基于 STAAR 开发出一条自动化分析流程,更好地推进技术成果落地。”谈及开发 STAARpipeline 的初衷,美国印第安纳大学医学院助理教授李子林表示。

图丨李子林(来源:李子林)
近年来,以二代测序技术为代表的高通量测序技术发展迅速,给精准医疗科研工作者提供了海量的基因组学数据。这些数据对揭示各种复杂疾病的遗传易感性及分子机制、识别高危个体或群体、发现防治干预靶点等具有重要意义。
因此,快速准确地分析规模化的基因组学和健康表型数据,并从中寻找致病基因、阐明基因与环境的交互作用,已成为全球精准医疗科研工作者关心的重要课题。
在此背景下,李子林所在的哈佛大学林希虹院士团队开发了罕见变异关联性分析工具 STAARpipeline。该工具拥有一体化关联分析流程,可以实现大规模全基因组数据分析流程的自动化,功能包括全基因组功能注释、常见变异和罕见变异关联分析、条件分析以及分析结果的汇总和可视化。
李子林说:“STAARpipeline 针对非编码基因组,提出了多种中罕见变异分析单位选择的新方法,显著地提高了检验功效,对非编码罕见变异分析有着重要的指导意义。”
2022 年 10 月 27 日,相关论文以《大规模全基因组测序研究中检测非编码罕见变异关联的框架》(A framework for detecting noncoding rare-variant associations of large-scale whole-genome sequencing studies)为题在 Nature Methods 上发表[1]。

图丨 相关论文(来源:Nature Methods)
李子林和哈佛大学陈曾熙公共卫生学院生物统计学系研究员厉希豪博士后担任共同一作,美国国家医学院院士、哈佛大学生物统计系和统计系教授林希虹和李子林担任共同通讯作者。
据李子林介绍,2018 年他加入林希虹团队,与美国国家心肺血液研究所的精准化医学研究计划(Trans-Omics Precision Medicine Program, TOPMed)团队合作,共同开展大规模全基因组测序数据的分析工作。
过程中他们发现,与基于阵列技术的全基因组关联研究数据不同,在测序数据中发现的变异位点,绝大部分都是罕见变异。为了更充分地利用测序数据中包含的信息,需要一个针对罕见变异的分析方法。
因此,在林希虹的指导下,李子林和其所在团队于 2019 年开发了非编码序列罕见变异关联性分析方法 SCANG[2]。2020 年,李子林和厉希豪带领团队开发了整合多组学数据的罕见变异关联性检验方法 STAAR[3]。而这两项工作,为 STAARpipeline 的开发,提供了坚实的基础。
后来,他们又发现,现有的罕见变异分析流程是孤立的,在采用上述工具分析数据时,还需要许多额外的步骤对数据进行预处理。这给医学和流行病学工作者带来了诸多不便。
同时,李子林还发现,目前的研究和方法大多聚焦编码基因组,忽视了全基因组中占比超过 98% 的非编码基因组。
基于此,李子林和厉希豪合作,对团队已有的统计分析方法进行整合,逐步实现了全基因组罕见变异关联分析工具的一体化和自动化,兼顾了统计的有效性、计算的可行性和可扩展性。
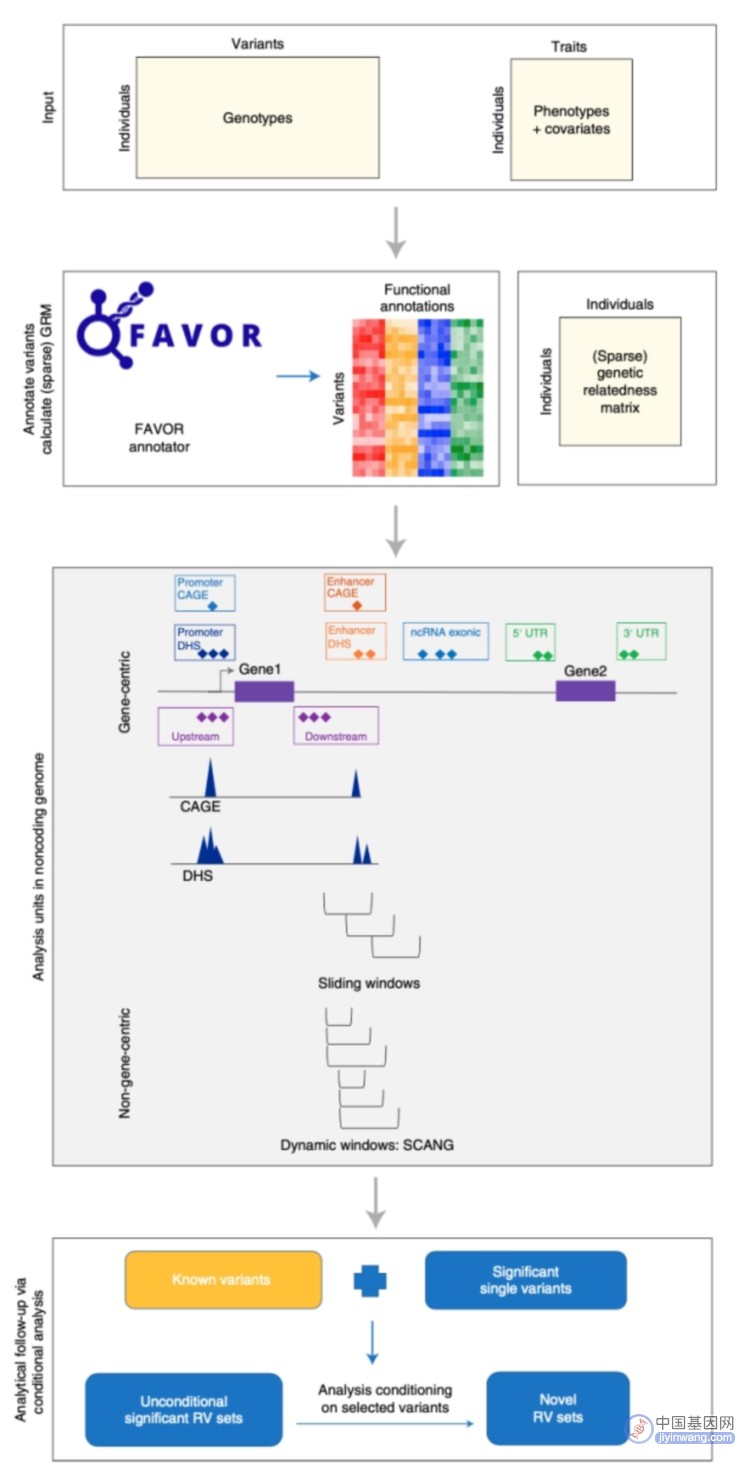
图丨 STAARpipeline 的工作流程(来源:Nature Methods)
“在软件开发的过程中,我们得到了 TOPMed 团队的大力支持。他们是我们的第一批用户,应用 STAARpipeline 分析了多种表型数据,并给予我们反馈,对工具的完善提出了很多有用的建议。”李子林表示。
他和团队其他成员把 STAARpipeline 应用到 TOPMed 全基因组测序数据中,高效分析了 40000 人的 9 种表型。“我们注意到,STAARpipeline 在以基因为中心的非编码分析中,发现了 49 个显著性关联,其中 35 个属于 6 个新的非编码功能类别集。”李子林说。
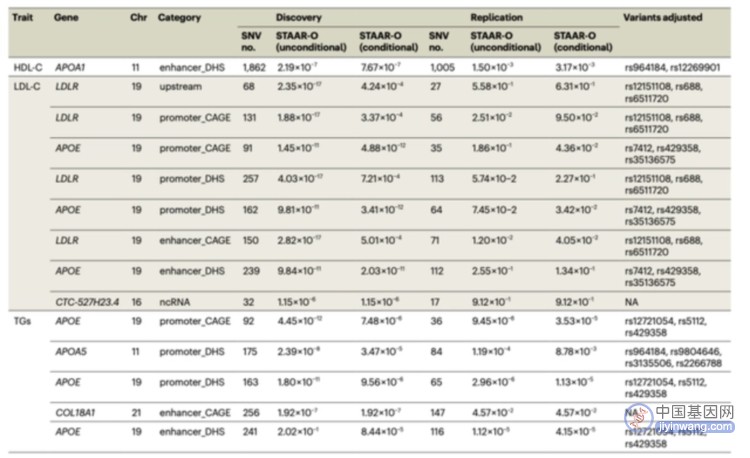
图丨以基因为中心的非编码分析结果(来源:Nature Methods)
目前,STAARpipeline 可以在高性能计算集群、云计算平台等多个运算平台上得到部署,帮助科研人员更好地分析大规模测序数据,并发现其中的致病基因和位点。
此外,李子林还介绍了关于该项研究的几个后续计划。
首先,其和团队将聚焦基因组大数据的统计分析方法研究,通过对基因组与代谢组、蛋白质组等多组学大数据的整合,探索罕见变异对疾病的影响。
其次,他们将继续开发和完善 STAARpipeline 的功能,逐步实现基因组大数据的标准化与可视化分析。
同时,该团队还会运用所研发的统计分析方法和工具,深入探究遗传变异与表型间的关系,致力于精准预防、精准医疗的研究和新的药物标靶的发现。
最后,李子林还表示,希望有机会和国内的团队合作,用开发的软件和工具分析国内的基因组大数据,尝试发现中国人群中的致病基因和位点,进而助力阐明可能的致病机制并开发有效的干预手段。
参考资料:
1. Li, Z., Li, X., Zhou, H. et al. A framework for detecting noncoding rare-variant associations of large-scale whole-genome sequencing studies. Nat Methods (2022). https://doi.org/10.1038/s41592-022-01640-x
2. Li Z, Li X, Liu Y, Shen J, Chen H, Zhou H, Morrison AC, Boerwinkle E, Lin X. Dynamic Scan Procedure for Detecting Rare-Variant Association Regions in Whole-Genome Sequencing Studies. Am J Hum Genet. 2019 May 2;104(5):802-814. https://pubmed.ncbi.nlm.nih.gov/30982610/
3. Chen H, Wang C, Conomos MP, Stilp AM, Li Z, Sofer T, Szpiro AA, Chen W, Brehm JM, Celedón JC, Redline S, Papanicolaou GJ, Thornton TA, Laurie CC, Rice K, Lin X. Control for Population Structure and Relatedness for Binary Traits in Genetic Association Studies via Logistic Mixed Models. Am J Hum Genet. 2016 Apr 7;98(4):653-66. https://pubmed.ncbi.nlm.nih.gov/27018471/
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





")















