Nature:人类基因组计划 探索未知领域的下一步
人类基因组计划(HGP)于1990年启动,旨在实现两个中心目标:1)分析人类 DNA 的结构;2)确定所有人类基因的位置。最近,我们已经成功实现了第一个目标,即获得了完整且连续的人类基因组DNA序列。然而,实现第二个目标比最初预期的要复杂得多,尽管我们对成千上万个人类基因的位置和功能有了更深入的了解。

那么,人类基因目录的现状如何?近年来为完成该目录所做了哪些努力?我们面临着哪些困难?最新一期的《自然》(Nature)杂志发表了一篇深入探讨这一问题的文章[1]。
基因组的早期概念将其视为基因的储存库,大多数基因被认为编码一个单一的蛋白质编码转录。然而,今天,我们知道情况不同了,人类的生物学可以受到成千上万的可选择的转录物和转录元素的影响,这些转录元素没有被翻译成蛋白质,还有成千上万的调节元素。更复杂的是,我们现在知道许多转录的RNA分子被进一步加工成更小的RNA片段,这些RNA片段的功能可能与它们的亲本转录本不同(图1)。
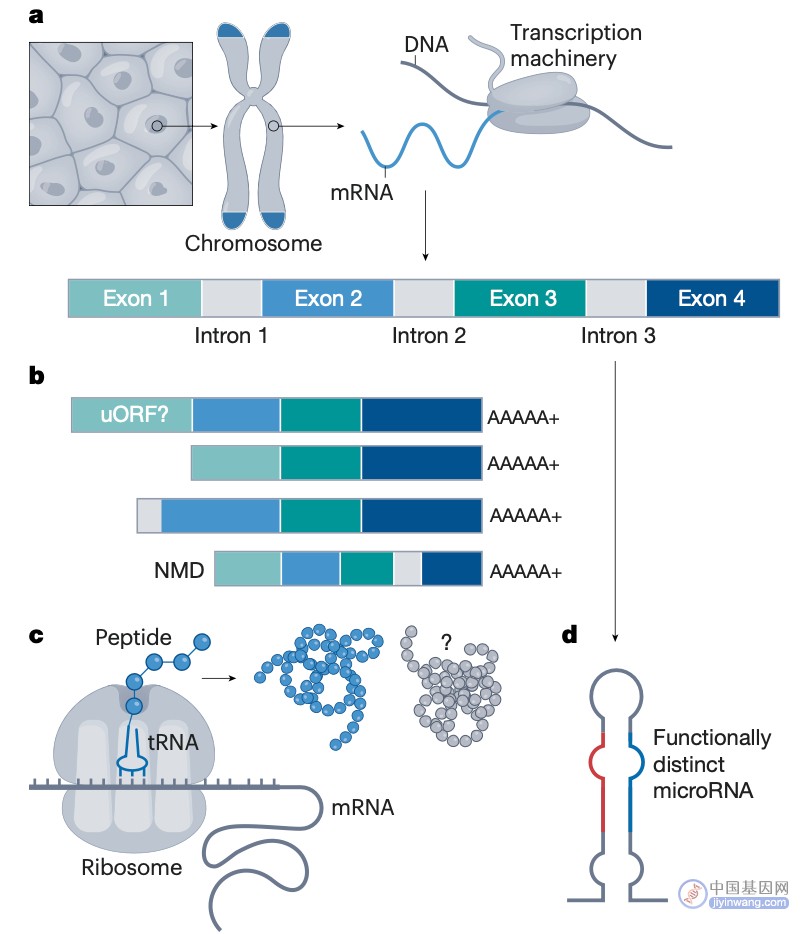
图1. 基因注释的一个主要挑战是如何捕获来自每个基因位点的基因产物的多样性。a,转录机制从细胞核中的 DNA 生成多个外显子和内含子组合,其中一个具有四个外显子(内含子/外显子比例未按比例显示)。b,转录产生的不同mRNA的样本。c,各种mRNA转录物从单基因座产生一种或多种不同的蛋白质。d,剪接内含子通常被细胞回收,但在某些情况下,它们编码功能不同的RNA种类,例如此处显示的从内含子3中出现的microRNA。
对此,来自多个国家的科学家们重新审视了人类基因组计划的目标,并深入探讨了在未来几年内完成人类基因注释的四项具体工作的进展、面临的挑战以及应对措施:
(1)完成蛋白质编码基因及其各种同工型的列表;
(2)编制包括各种长度和类型的完整RNA基因清单;
(3)识别与医学上重要的基因和基因变异相关联的特定疾病,并建立联系;
(4)完善实现人类基因注释所需的技术。
第一项工作:蛋白质编码基因注释的进展与挑战
蛋白质编码基因的注释是人类基因组计划的主要焦点,在捕获序列本身之后,虽然这种注释仍然不完整,但科学界正在接近对这些基因的身份达成共识。
从20世纪80年代最初估计的5万到10万个基因,基因的估计数字稳步下降,随着人类基因组的首次发表,下降到3万到4万个,然后进一步下降到25,000,今天略低于2万个,最近的一个数据库发布表明只有1.9万个基因(例如,GENCODE第41版的19,370个)。这些数据的调整来自于各种各样的进步,包括全面的人工审查,计算注释方法和分析的改进,以及越来越多的高质量实验转录数据的产生。尽管基因数量总体减少,新的蛋白质编码基因继续被鉴定,以及已知基因的替代同种异构体。
MANE(Matched Annotation from the NCBI and EMBL-EBI,Ensembl/GENCODE1和RefSeq发起了一项联合倡议,以汇集人类基因和转录物注释,并共同定义一组高价值的转录物和相应的蛋白质)合作组织最近发布了一个近乎完整的数据集,其中包含每个蛋白质编码基因的一个同种异构体,这两个领先的注释项目RefSeq和GENCODE完全同意。MANE 1.0包含19062个基因位点,占人类主要基因目录中蛋白质编码位点总数的95%。这个正在进行的项目有望为我们有多少蛋白质编码基因这个问题提供一个明确的答案。
研究者还提出了一些完成人类基因组中蛋白质编码基因注释的未来步骤(挑战):
1)对于每个蛋白质编码基因,开发其转录物及其在所有可用组织和细胞类型中的表达水平的综合图片,并确定其在其他物种中的保守性。
2)对于所有折叠成稳定结构的蛋白质,确定其三维结构并评估其稳定性。
3)确定转录起始和终止的所有可选位点,并记录每个位点在正常组织中被利用的频率。
4)标记所有导致无功能蛋白的可重复剪接事件。
5)列出并突出显示违反正常规则的许多例外情况。
第二项工作:非编码RNA基因的注释的进展与挑战
非编码RNA基因(ncRNAs)是一类从DNA转录而来的RNA分子,不编码蛋白质,但在细胞中发挥功能。然而,确定哪些RNA具有功能性是注释工作的主要挑战之一。尽管许多非功能性RNA序列可能在多种细胞和条件下被转录,但我们只将具有可识别功能的RNA称为基因。目前,大部分注释工作集中于全面编目ncRNA转录物,而不考虑其功能状态。
对ncRNA进行标注的一个主要挑战是添加功能标签。对于蛋白质编码基因而言,我们拥有丰富的先验功能证据,并且可以依靠基于一级序列的强大计算方法预测基因功能。例如,通过翻译的氨基酸序列,我们通常可以预测DNA结合转录因子或膜结合受体。然而,对于大多数ncRNA,我们仍了解甚少,也没有有效的序列预测功能方法。因此,近期注释ncRNA基因的目标之一是描述支持它们的不同类型证据,例如组织特异性表达水平,尽管它们的功能可能仍然未知。
第三项工作:基因注释的进展与挑战
人类基因注释在遗传病的诊断和治疗中具有重要应用。根据OMIM49的编目,有5000多个基因和数千个变异与单基因失调和疾病风险相关。例如,BRCA Exchange数据库(https://brcaexchange.org/) 目前仅列出了BRCA1基因的34,000多个变体,其中2228个被标记为致病性。
在评估变异的致病性时,基因和转录本模型的完整性和准确性至关重要。使用PolyPhen51、Revel51和Variant Effect Predictor (VEP52)等工具确定的变异影响取决于预测的开放阅读框转录本。此外,临床诊断分析中使用的靶向捕获测序的寡核苷酸诱饵和PCR引物的设计依赖于正确的外显子边界注释。即使使用全基因组测序(WGS)进行诊断,临床医生也不会考虑将未注释的外显子作为解释的候选。
这一领域面临的主要挑战在于:需要一个临床标准。目前,临床实验室通常在GRCh37 (hg19)人类组装上操作,并使用RefSeq转录本作为已知与疾病相关的基因的参考,通常依赖于文献报道。当文献不明确时,实验室倾向于使用简单的标准来选择转录本,例如长度或在注释数据库中的首次出现。然而,这种做法存在两个主要问题:首先,并非所有的RefSeq转录本都能完美地映射到GRCh37人类参考基因组;其次,所选的转录本可能无法反映临床诊断所需的特征或最具代表性的转录本。
第四项工作:创新技术的应用挑战
未来基因目录的完成需要创新的新技术来解决未来的挑战。
基因产物的匹配可以通过长读测序和蛋白质组学分析来实现。目前,需要对特定同种异构体的时间和位置进行全基因组测量。在组织内和单细胞分辨率上测量基因表达已经揭示了细胞和组织中基因表达的许多协调模式。然而,目前存在着细胞特异性剪接估计的问题,剪接事件的数量可能被低估了。同种异构体水平的RNA-seq分析目前依赖于基因内外显子的差异表达,这在很大程度上受到文库构建方法和测序深度的影响。即使准确测量了表达水平,转录物的相对丰度也不能完全与翻译相关。
捕获低表达转录本的方法也是一个重要的创新。捕获测序最近已经被应用于靶向特定RNA,以便以高通量方式使用短读和长读RNA测序为基因组的选定区域提供更高的测序覆盖率。这对于从超低输入样本和极低表达水平的基因中富集RNA特别有用。捕获技术的使用,结合最近长读测序平台吞吐量的增加,可以极大地有利于低表达转录物的研究,特别是长非编码RNA(lncRNA),这对于正常和病变细胞的基因调控研究至关重要。
总结
在人类基因组最初发表的20多年后,蛋白质编码基因的数量稳定在19,500左右,尽管这些基因的同种异构体的数量仍然是一个深入研究和讨论的主题。人类基因组序列的完成本身就提供了将这些基因映射到一个稳定的、完整的序列上的机会,并在未来几年内收敛到最终数量。基因和异构体注释的更大标准化将提高我们在临床环境中应用这些知识的能力。
相比之下,非编码RNA基因,特别是lncRNA,处于早期认识阶段,数量仍在增加,目前的目录包含17,000-20,000个或更多的lncRNA。新技术为完善这一目录提供了有希望的途径,尽管lncRNA的完整功能表征可能还需要许多年的时间。在过去的20年里,蛋白质编码基因的数量稳步下降(图2),随着我们对RNA生物学和技术的了解的提高,lncRNA的数量是否会遵循类似的趋势,这是很自然的问题。
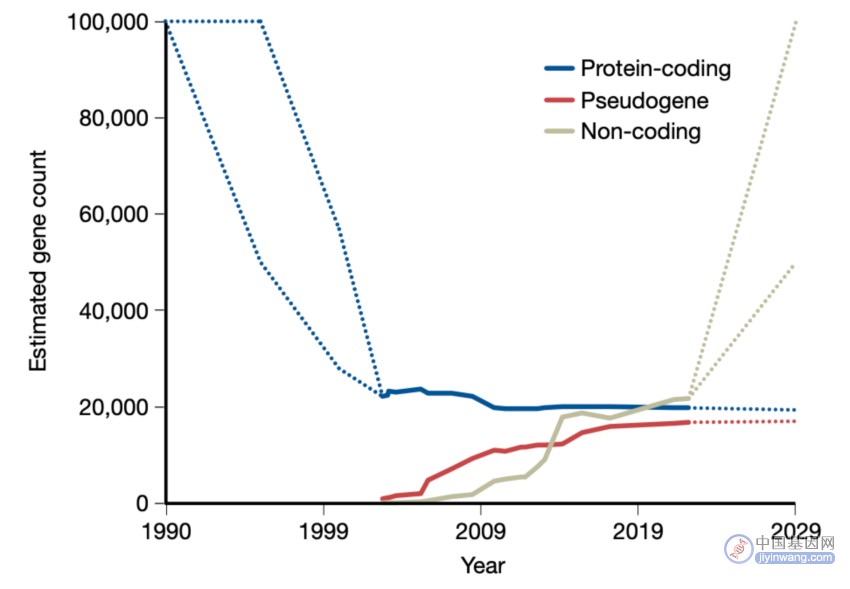
图2. 预测和观察到的人类基因计数随时间的变化。显示了蛋白质编码基因、假基因和非编码基因的计数。2003年之前和2023年之后的时间点(虚线)分别代表了文献预测的平均值和从这个角度推断的平均值。
当然,我们也注意到,即使完成了基因组的完整基因注释,我们也只有一个人类基因目录的例子,这个例子并不适用于所有人类。很可能许多健康的个体都有或多或少的某些基因拷贝,未来调查人类种群多样性的努力将是实现更完整的基因组基因内容视图的重要一步。
参考文献:
1.Amaral P, Carbonell-Sala S, De La Vega FM, et al. The status of the human gene catalogue. Nature. 2023 Oct;622(7981):41-47. doi: 10.1038/s41586-023-06490-x.
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。






![基因测序行业深度研究报告:未来大健康领域黄金赛道[共77页]](/static/upload/other/20230126/1674744324488315.jpeg "基因测序行业深度研究报告:未来大健康领域黄金赛道[共77页]")














