《自然》揭秘癌症最强基因RAS促癌新机制!使干细胞和微环境沟通混乱
鳞状细胞癌起源于上皮层,是一种常见的癌症病理类型,多见于肺癌、食管癌及口腔癌。肺鳞癌占所有肺癌患者的30%-35%[1],在食管癌中的鳞癌占比更是高达90%[2]。近年来,由于环境因素的改变,鳞癌发病率越来越高[3]。
那究竟是什么因素驱动了鳞癌的发生呢?
研究表明,外在环境中的化学致癌因素可通过诱导RAS突变以及激活RAS-MAPK信号通路促进鳞癌的发生[4]。然而,RAS突变驱动癌症发生的内在机制仍不清楚。
近日,由洛克菲勒大学的Elaine Fuchs教授领衔的研究团队,在《自然》杂志发表了一项重要研究成果。
他们通过单细胞RNA测序深度解析了鳞癌干细胞的特征,发现在致癌RAS启动后,组织干细胞与周围环境会出现“错误沟通”,导致血管重塑及TGF-β信号通路传导,进而促进基质瘦素受体异常表达和PI3K-AKT-mTOR的异常激活,从而促进组织由良性到恶性的转变[4]。
总之,这项研究揭示了RAS遗传性突变可通过干细胞与微环境的异常通讯导致致癌信号通路的激活,从而促进鳞癌发生。

RAS突变的皮肤干细胞进展为侵袭性鳞癌,需要经历良性肿瘤这一过程。
为模拟鳞癌发生过程,Fuchs团队通过胚胎注射慢病毒构建RAS基因突变鼠(TRE-HRASG12V),在此基础上使用四环素诱导皮肤鳞癌小鼠。他们发现,在第4周时可诱导出增生性的良性肿瘤,而在第8周即可进展为未分化的浸润性鳞癌。
此外,他们通过标记皮肤干细胞发现,相比于良性肿瘤,鳞癌中的干细胞存在明显的TGF-β信号通路及下游转录因子SMAD2激活。
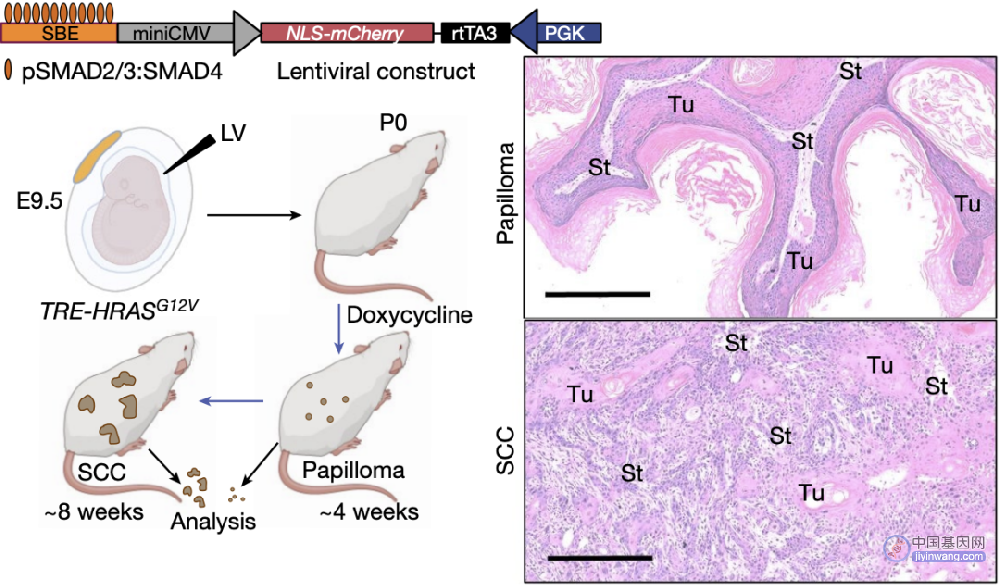
鳞癌小鼠模型的诱导
为明确RAS介导的干细胞癌变的机制,他们对鳞癌组织进行了单细胞RNA测序分析,根据测序结果将细胞分为3簇(C1、C2和C3),并发现癌症干细胞标志物CD200、HMGA2和PTHLH在C2簇中显著富集。
为进一步探明干细胞癌变的分子特征,他们对C2簇细胞的特征性基因进行了聚类分析,发现血管生成、细胞迁移、伤口愈合、蛋白质磷酸化和胞内信号转导等途径显著富集(图3D)。随后,他们标记了鳞癌和良性肿瘤组织中C2簇细胞标志物Keratin18和血管生成标志物CD31,以检测良性肿瘤和鳞癌组织血管生成的差异,发现在Keratin18阳性的鳞癌和良性肿瘤组织中,鳞癌组织中CD31的表达显著高于良性肿瘤。
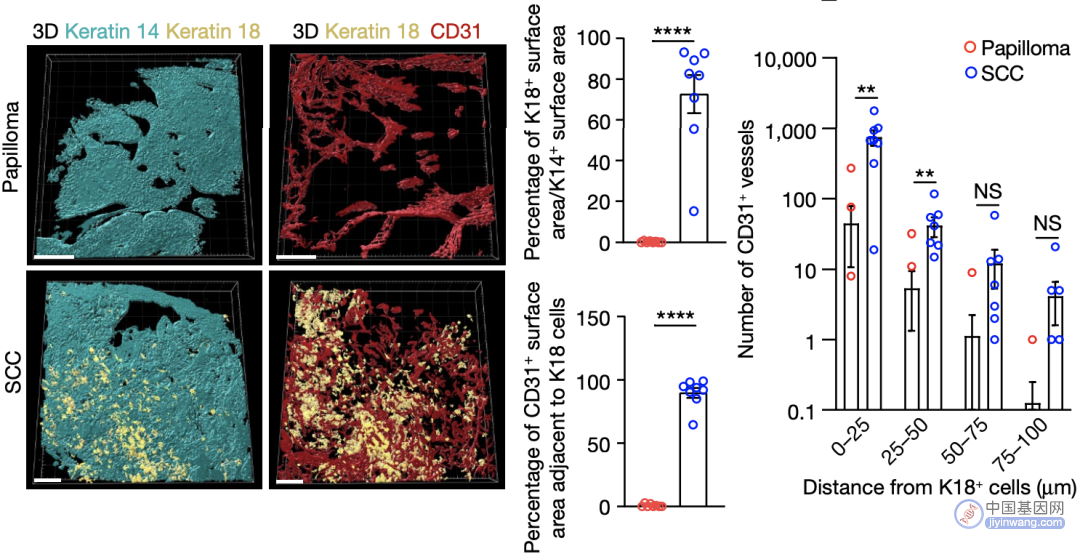
良性肿瘤与鳞癌组织CD31的表达
由此可见,正常皮肤干细胞癌变的过程中会同时出现TGF-β信号通路的激活以及血管生成的增强。此外,在TGF-β信号通路上调的C2簇细胞中,与鳞癌干细胞自我更新和增殖、基底膜重塑、细胞迁移、血管生成相关的101个基因也显著上调。这表明在鳞癌发生过程中,干细胞、TGF-β介导的信号通路和血管生成三种因素出现了相互作用。
上述101个基因中,有43个基因在由良性肿瘤向鳞癌演变的过程中特异性上调,瘦素受体(LEPR)引起了Fuchs团队的注意。
一般情况下,LEPR仅于肥胖小鼠中表达,在鳞癌模式鼠这种营养健康的小鼠中几乎不表达,但他们发现LEPR在TGF-β信号通路上调的鳞癌干细胞中显著上调,这就有些反常了。
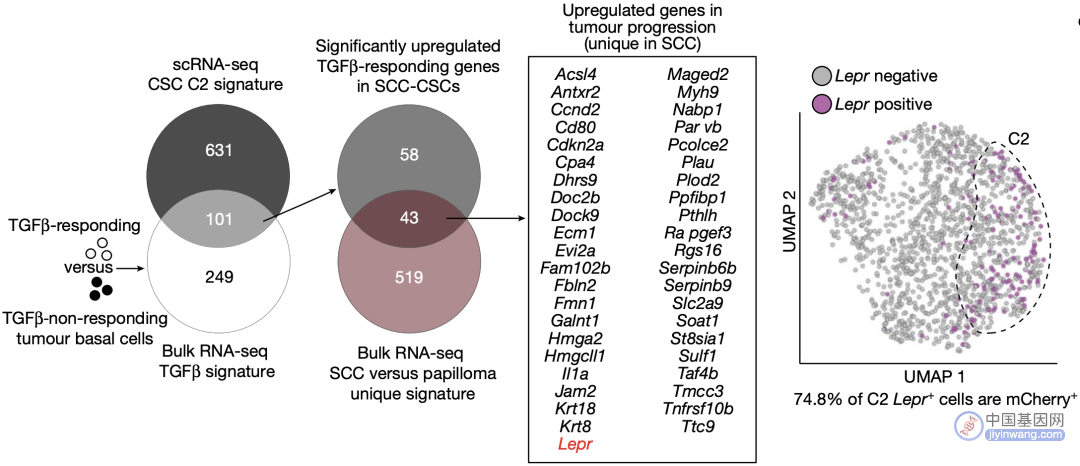
LEPR在TGF-β信号通路上调的鳞癌干细胞中显著过表达
为探明LEPR是否参与了皮肤干细胞癌变的过程,Fuchs团队首先通过集落形成实验及裸鼠荷瘤实验检测了LEPR对细胞干性和鳞癌发生的影响,结果表明,LEPR的缺失可使鳞癌细胞集落形成能力下降3倍(C3 vs C2),裸鼠成瘤能力下降10倍。
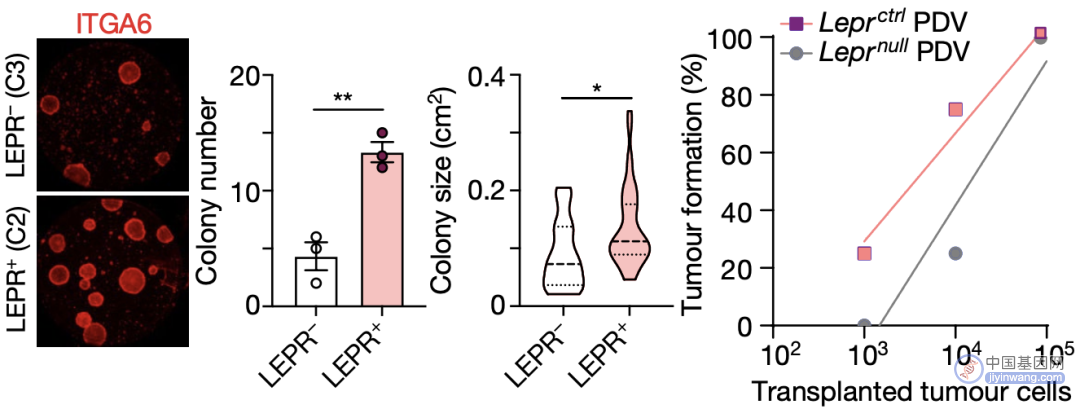
LEPR对细胞干性和成瘤能力的影响
Fuchs团队还发现,鳞癌中的瘦素水平高出良性肿瘤组织5倍以上,但良性肿瘤和鳞癌细胞中均无瘦素配体,所以这种瘦素的升高很有可能是由于异常的肿瘤微环境。因此,他们提出了两种LEPR升高的假说:(1)局部脂肪输送;(2)血管生成增加,瘦素从血管渗透入肿瘤组织。
经验证,肿瘤微环境中无明显脂肪积累,但通过向小鼠皮下注射血管内皮生长因子A(VEGFA)促进血管生成可导致肿瘤生长加速和瘦素水平上调(图6),因此,肿瘤微环境中瘦素水平上调和血管生成增加有着紧密联系。
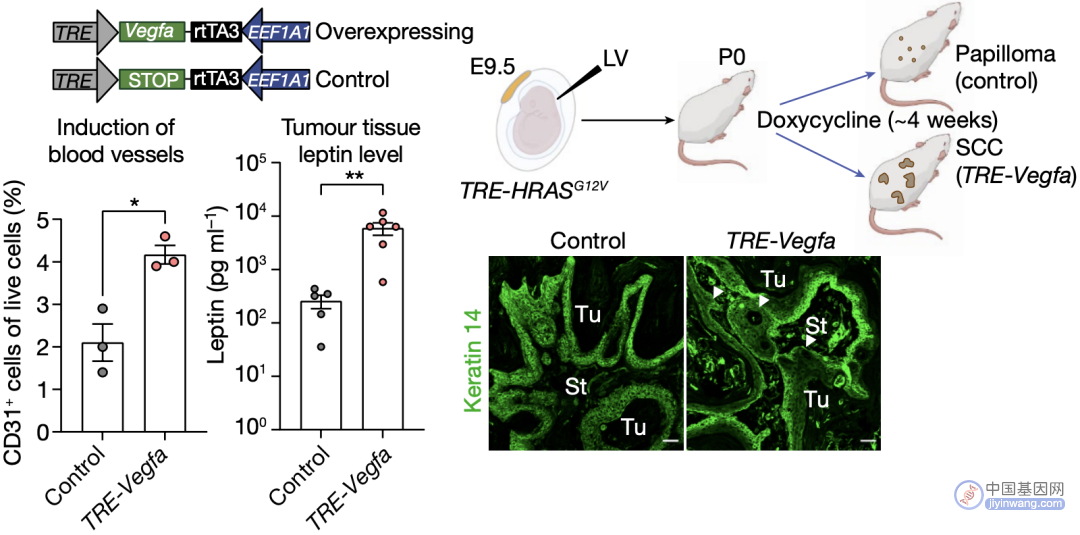
血管生成增加促进血管生成、肿瘤生长和瘦素水平上调
已有研究表明,LEPR可促进经典致癌通路PI3K-AKT-mTOR的激活[5],且鳞癌细胞转录组KEGG分析发现LEPR与中PI3K-AKT信号通路呈正相关。
为进一步明确鳞癌中LEPR与PI3K-AKT信号通路的调控关系,研究人员敲除了鳞癌细胞中的LEPR,发现LEPR的缺失显著下调了AKT的磷酸化,且PI3K抑制剂BKM120可显著抑制LEPR表达的肿瘤,但对LEPR缺失的肿瘤影响不大。
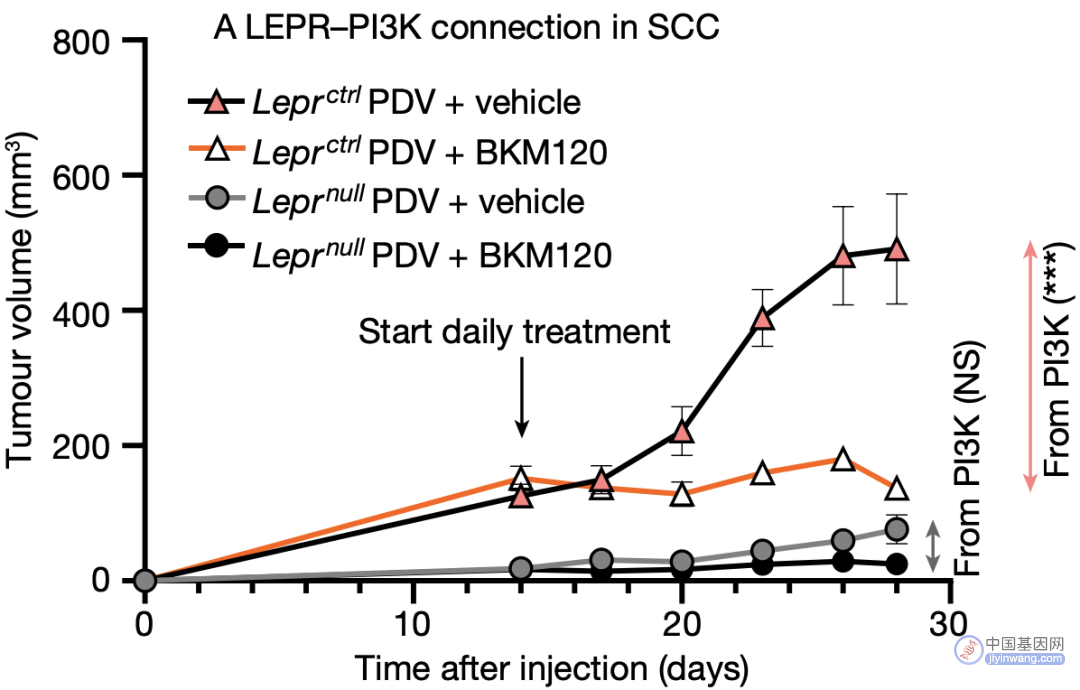
LEPR对AKT信号的影响
此外,与LEPR表达的肿瘤组织相比,LEPR缺失的肿瘤组织中mTOR的关键蛋白pS6显著下调,且mTOR抑制剂雷帕霉素可显著抑制LEPR表达鳞癌的生长,但对LEPR缺陷的肿瘤影响较小。
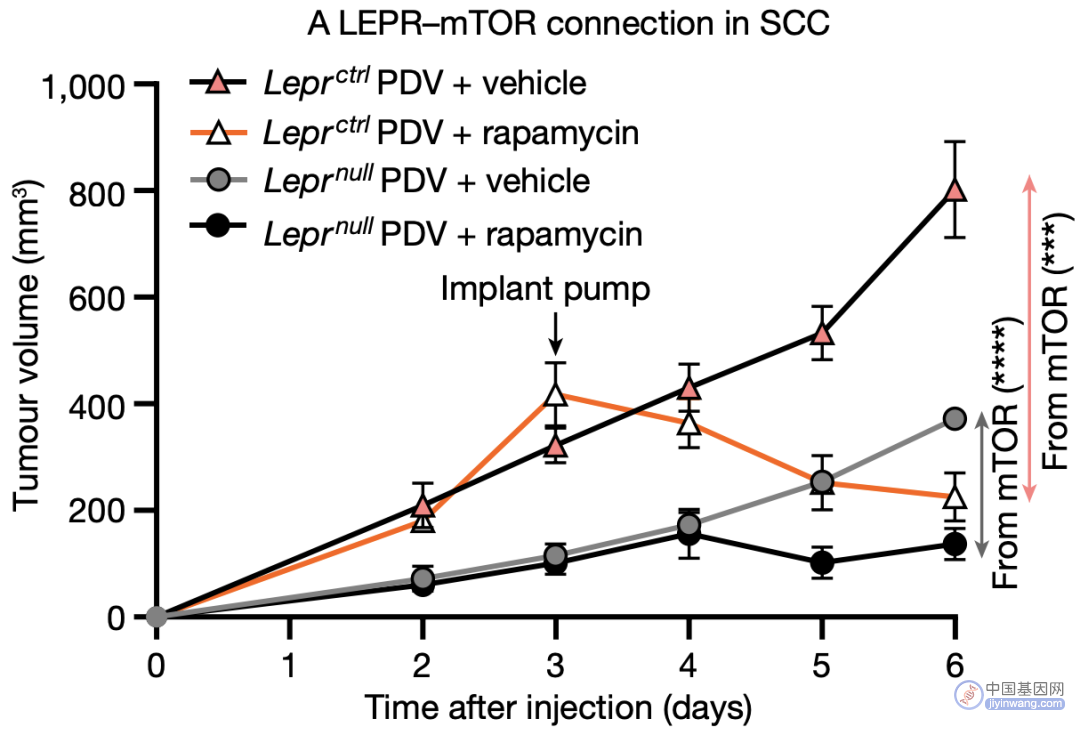
LEPR对mTOR信号的影响
由此可见,LEPR可能是通过激活PI3K-AKT-mTOR信号通路,进而促进了鳞癌发生发展。
总的来说,这项研究揭示了一种致癌基因突变通过动态影响肿瘤微环境,促进癌症发生的独特机制。具体来说,RAS突变可通过诱导鳞癌干细胞与微环境的相互作用,促进微环境中血管重塑和LEPR过度表达,导致PI3K-AKT-mTOR信号通路的激活,进而促进鳞癌发生。
此外,这项研究成果也为LEPR/PI3K通路抑制剂的开发和临床转化,提供了理论依据。
参考文献
[1] Lau SCM, Pan Y, Velcheti V, et al. Squamous cell lung cancer: Current landscape and future therapeutic options. Cancer Cell. 2022 Nov 14;40(11):1279-1293. doi: 10.1016/j.ccell.2022.09.018.
[2] Thrift AP. Global burden and epidemiology of Barrett oesophagus and oesophageal cancer. Nat Rev Gastroenterol Hepatol. 2021 Jun;18(6):432-443. doi: 10.1038/s41575-021-00419-3.
[3] Dotto GP, Rustgi AK. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell. 2016 May 9;29(5):622-637. doi: 10.1016/j.ccell.2016.04.004.
[4] Yuan S, Stewart KS, Yang Y, et al. Ras drives malignancy through stem cell crosstalk with the microenvironment. Nature. 2022 Nov 30. doi: 10.1038/s41586-022-05475-6.
[5] Friedman J. The long road to leptin. J Clin Invest. 2016 Dec 1;126(12):4727-4734. doi: 10.1172/JCI91578.
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





")












")


