诺禾致源:组装新策略完成犬蝠基因组精细图谱并揭示食果蝙蝠免疫基因演化新机制
近日,国际TOP期刊Science Advances在线发表了题为“Comparative analyses of bat genomes identify distinct evolution of immunity in Old World fruit bats”的研究论文。该研究由国内外多家单位研究人员共同协作完成。北京诺禾致源科技股份有限公司首席科学家、武汉大学田仕林博士和武汉大学曾嘉鸣为该论文的共同第一作者。
图1 来自Science官网
该研究提出了一种改良的四步法基因组组装策略,完成了一个近乎完整的染色体级别的狐蝠科犬蝠属犬蝠(Cynopterus sphinx)全基因组序列图谱,并结合比较基因组学与细胞学实验揭示了狐蝠科蝙蝠免疫基因演化的新机制,提示食果蝙蝠在应对病毒感染时可能有独特的免疫机制。
蝙蝠已经演化出独特的免疫系统来应对病毒感染,能够无症状地携带多种哺乳动物烈性病毒,包括尼帕病毒、亨德拉病毒、埃博拉病毒、马尔堡病毒、MERS病毒、SARS病毒以及最近引起COVID - 19的SARS - CoV - 2病毒,是这些人畜共患病毒的天然宿主。狐蝠科蝙蝠(旧大陆果蝠)被认为是引起国际公共卫生组织关注的多次病毒大流行事件中高致病性病毒的天然宿主。因此,研究狐蝠科蝙蝠免疫系统演化遗传机制对人类制定病毒的防御和抗病毒治疗策略具有重要的意义。
拓展阅读
研究结果
改良的四步法基因组组装策略完成了犬蝠高质量参考基因组
该研究提出应用“先全局组装,再聚类长读长reads,接着局部组装,后染色体挂载”的四步法改良基因组组装策略,不同于当前常规的两步法组装流程(先组装后染色体挂载)。该流程利用了染色质互作数据(Hi-C)预先对具有潜在连接关系的Nanopore长序列进行聚类,有效地去除了组装过程中构建序列串图(string graph)时的远距离重复序列的嵌合连接。详细步骤如下(图2A):
(1)组装和纠正重叠群(Contigs):基于高质量的三代测序数据,应用通用的构建序列串图(string graph)组装方法完成初始的Contig序列的组装,然后利用基因组纠错软件来对初始Contig进行纠错。从而得到了基因组序列Contig v1。
(2)通过Hi-C测序数据的互作频率对Contig v1聚类:首先使用Bowtie2 软件的单端模型将Hi-C测序数据映射到Contig v1上;接着,筛选出唯一的映射对后,使用 HiCUP流程过滤掉无效的自连接和未连接片段;然后,使用有效地互作reads使用凝聚层次聚类算法计算所有Contigs之间的链接频率。最后,根据Contig之间的互作信号的密度大小判定聚类,并结合组装物种的核型数据预设染色体连锁群数目。通过上述操作,得到基因组的Contig连锁群。
(3)对三代测序数据进行比对分类以执行连锁群局部组装:首先使用 Minimap2软件将三代测序数据重新对齐到 Contig v1。在去除次优比对reads后,提取了每个连锁群中最优比对的三代测序序列。接着,对每个连锁群中比对的三代测序序列执行连锁群局部组装。与全局组装相比,该策略可以有效地避免在组装过程中构建string graph时,由于基因组远距离重复序列引起的错误重叠关系。然后,评估每个连锁群局部组装的适当组装参数,随后进行基因组组装和校正,方法与步骤(1)中提到的类似。最后,获得了第二个基因组序列 Contig v2。
(4)将Contigs锚定到染色体上:利用ALLHiC软件计算Contig v2中序列之间的互作频率挂载基因组的染色体序列。随后,将Hi-C互作频率信息输入到Juicebox软件中进行可视化,再通过手动调整互作频率表现出明显离散的Contig之间的错误位置和方向。
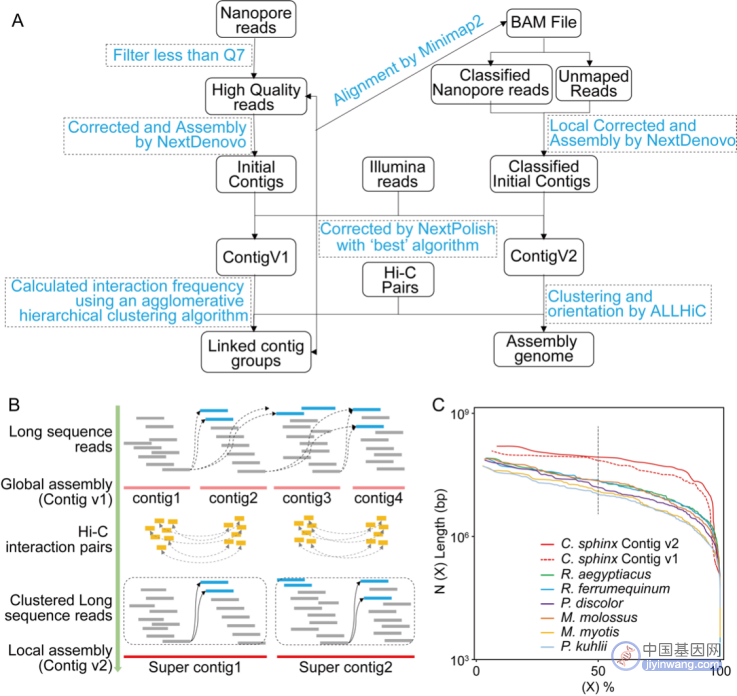
图2 改良的基因组组装策略
应用上述的四步法改良基因组组装策略(图2),完成了一个近乎完整的染色体级别的狐蝠科(旧大陆果蝠)犬蝠属犬蝠基因组序列图谱。相比于传统的两步法,在基因组的连续性、完整性和准确性都表现出了优秀的性能。特别是,应用本文中的四步法组装策略,完成了4条T2T级别的染色体序列。该方法还具有改良现有基因组组装质量的潜力,比如,在本文中,应用四步法组装策略,重新将已发表的库氏伏翼蝠,(P . kuhli,论文比较基因组研究物种之一)的不完整X染色体组装完全。
狐蝠科蝙蝠基因组进化
对11种蝙蝠(其中包括6种狐蝠科蝙蝠)和7种模式哺乳动物进行进化树分析,发现蝙蝠阴翼手亚目和阳翼手亚目分化于65.5 MYA(图3A)。之后分析了17个哺乳动物基因组与C . sphinx基因组的共线性,发现随着与C . sphinx 进化距离的增加,共线性比率降低,并且狐蝠科内的物种之间具有更高水平的共线性。比较基因组分析发现C . sphinx与其他蝙蝠之间的大型染色体间重排事件(长度 ≥ 1Mb)主要发生在C . sphinx中最长的前12条染色体上,除了LG10(图3B)。进一步分析发现C . sphinx LG10染色体序列与非蝙蝠哺乳动物的X染色体序列唯一匹配,平均共线性率为67.12 % (从小鼠的51.84 %到马的74.05 %),因此推测LG10为C . sphinx的X染色体。值得注意的是,在所有蝙蝠的基因组中,除了P . kuhli外,都有一条染色体与C . sphinx的X染色体具有良好的共线性(从M. molossus的69.76 %到R . aegyptiacus的87.41 %)。推测已发表的P . kuhli X染色体序列被组装成了多个独立的Scaffold序列,因此,应用本论文中提出四步法组装策略重新组装了一条完整的P . kuhlii X染色体序列。总之,研究结果表明了蝙蝠的X染色体与其他哺乳动物一样,是高度保守的(图3)。
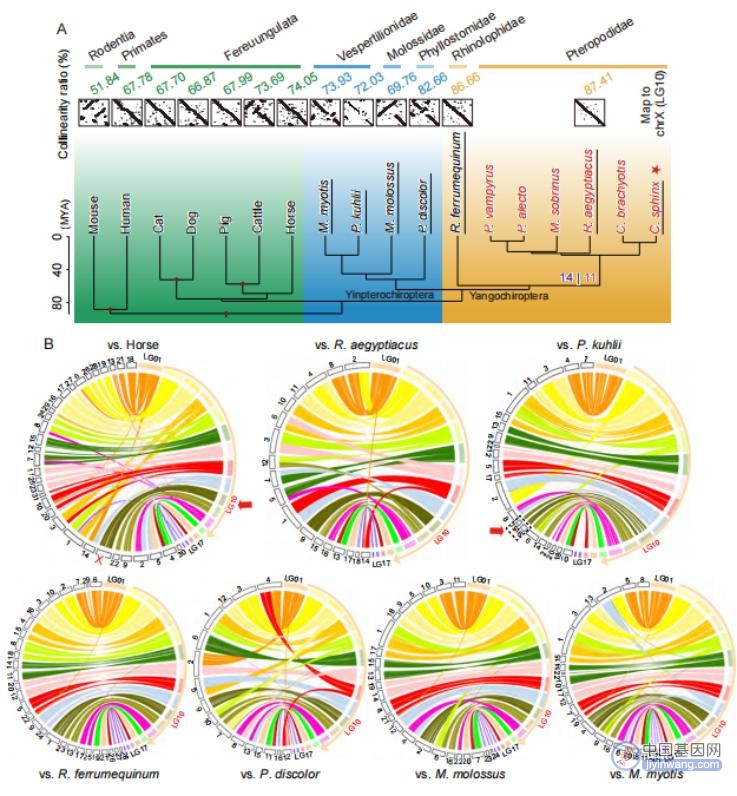
图3 进化树和基因组共线性分析
论文中还以狐蝠科染色体级别的基因组C . sphinx和R . aegyptiacus为代表,进行了特异性序列鉴定和分析,结果发现狐蝠科基因组中携带内源性逆转录病毒元件片段。这些内源性逆转录病毒大多数属于γ逆转录病毒,其次是β逆转录病毒。对每个逆转录病毒基因的5 和3 长末端重复序列(long terminal repeat,LTR)进行预测并估计它们在逆转录病毒感染时的分歧率。结果发现这两个狐蝠科都经历了两个古老的逆转录病毒整合爆发事件,一个在1 - 3 Mya左右,另一个在10 - 12 Mya左右。由于蝙蝠比大多数其他哺乳动物更耐受病毒感染,因此推测更频繁的病毒基因整合到宿主基因组中可能是狐蝠科基因组多样性的重要因素之一。
狐蝠科基因家族进化
基因组片段复制(Segmental duplications,SDs)是基因组大片段(> 1 kb)和高一致性(> 90 %序列一致性)区域拷贝数倍增事件,对基因组中的基因创新具有重要意义。因为SD序列具有高重复性,往往对于基因组组装带来高难度挑战。因此,该研究只对于7种染色体级别的蝙蝠基因组进行了SD分析,平均鉴定了34.38 Mb的SDs,含有611个基因。论文在两只狐蝠科蝙蝠(C. sphinx和R . aegyptiacus)的特有SD区域鉴定到24个人类同源基因,其中20个至少在一个狐蝠科基因组中发生了基因复制。其中包括一个肽聚糖识别蛋白(peptidoglycan recognition protein,PGRP)家族成员PGLYRP1,该位点在哺乳动物中一般是单拷贝形式,其蛋白产物由活化的中性粒细胞释放,通过与肽聚糖结合识别感染的微生物,发挥抑菌和杀菌活性。因此,推测狐蝠科蝙蝠中该基因拷贝数变异有助于减轻其炎症反应。
为进一步解析狐蝠科中基因家族的进化关系,将鉴定到的基因家族分为核心(跨所有物种共享)的基因家族(67.68%),部分共享的基因家族(25.58%)和物种特异性的基因家族(6.75%)(图4A)。对狐蝠科扩张和收缩的基因家族进行分析,共鉴定到14个扩张的基因家族和11个收缩的基因家族。有趣的是,大多数扩张的基因家族中至少有一个位于C . sphinx和R . aegyptiacus基因组的SD区域内,再次表明SDs可能与基因组中的基因创新有关。
论文在狐蝠科收缩的基因集中,发现NLR基因(nucleotide-binding domain leucine-rich repeat)家族中的关键炎症小体NLRP1基因在所有狐蝠科蝙蝠中缺失(图4B)。NLRP1是第一个被证明可形成炎症小体的蛋白。通过比较蝙蝠中NLRP1(或其推断的删除位点)上下游同源的重复序列类型LINE - 1(L1)插入情况,发现狐蝠科中L1插入数量和累积长度均较低。因此,推测L1插入介导NLRP1基因序列的缺失,从而导致相关炎症反应的减弱。在狐蝠科蝙蝠扩张的基因家族中,发现了C5AR2基因序列的串联重复(图4C)。C5AR2是补体过敏毒素C5a的受体,具有调节TLR4介导的NLRP3炎症小体活化和巨噬细胞IL1 β释放的抗炎功能。与5个非狐蝠科蝙蝠中发现的C5AR2单拷贝不同,在狐蝠科中发现了2个拷贝,并且位于基因组SD区域。论文推测C5AR2基因的串联重复可能增强了狐蝠科巨噬细胞IL1 β释放的能力,从而抑制病毒感染后的炎症反应。
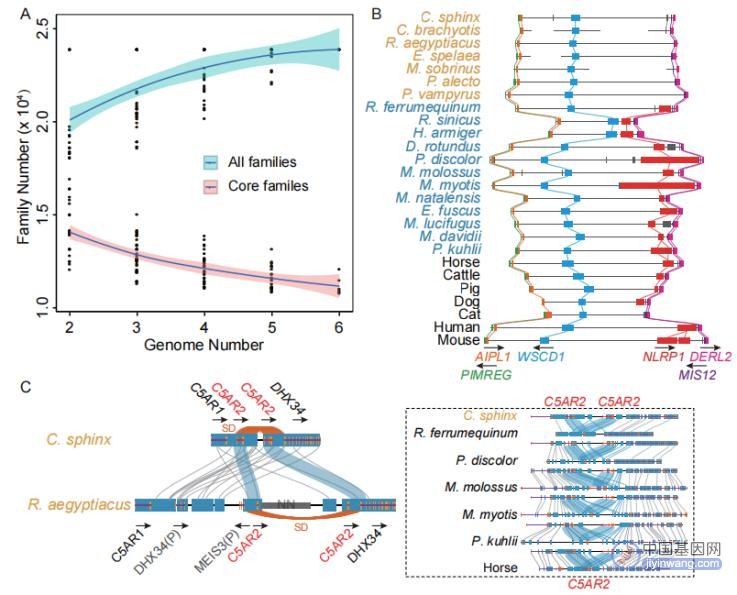
图4 基因家族进化分析
狐蝠科蝙蝠免疫基因进化
通过对狐蝠科的祖先分支应用PAML中的分支测试和分支位点测试,分别鉴定到316个快速进化基因(REGs)和241个正选择基因(PSGs)。在这些基因中,分别有35个REGs (11 %)和46个PSGs (19 %)在免疫和/或抗病毒反应中发挥作用。而对REGs和PSGs富集分析发现狐蝠科进化出了不同的适应机制来对抗病毒感染,包括模式识别和干扰素(IFN)介导病毒复制(图5A)。研究还发现了狐蝠科正选择基因IGF2F(胰岛素样生长因子2受体)在宿主防御的第一道防线中发挥作用;狐蝠科正选择基因MEFV作为自噬受体降解多个炎性小体成分(如NLRP3),为之前证明的蝙蝠NLRP3炎性小体活化水平低提供了新的证据;狐蝠科正选择基因环状GMP-AMP合成酶基因cGAS的N端和C端序列的特异性分子改变分别表现出增强对病毒DNA感知和减弱病毒炎症反应的不同免疫功能。
论文还通过同源比对搜索了KOBAS 3.0数据库中与免疫系统过程和病毒过程相关的直系同源基因,一共鉴定了2,162个免疫相关的直系同源基因集。利用PAML软件包的one-ratio模型(model 0)证明了狐蝠科蝙蝠的平均ω值显著地高于其它蝙蝠和非蝙蝠哺乳动物(图5B)。还通过比较模型M8a(ω = 1)与模型M8(ω > 1)对每个免疫基因进行正选择位点模型测试,发现狐蝠科蝙蝠的正选择基因数目也极显著多于其它蝙蝠和非蝙蝠哺乳动物(图5C)。因此,论文得出了狐蝠科蝙蝠在免疫相关基因中表现出比其它蝙蝠和非蝙蝠哺乳动物更高的分子适应率。
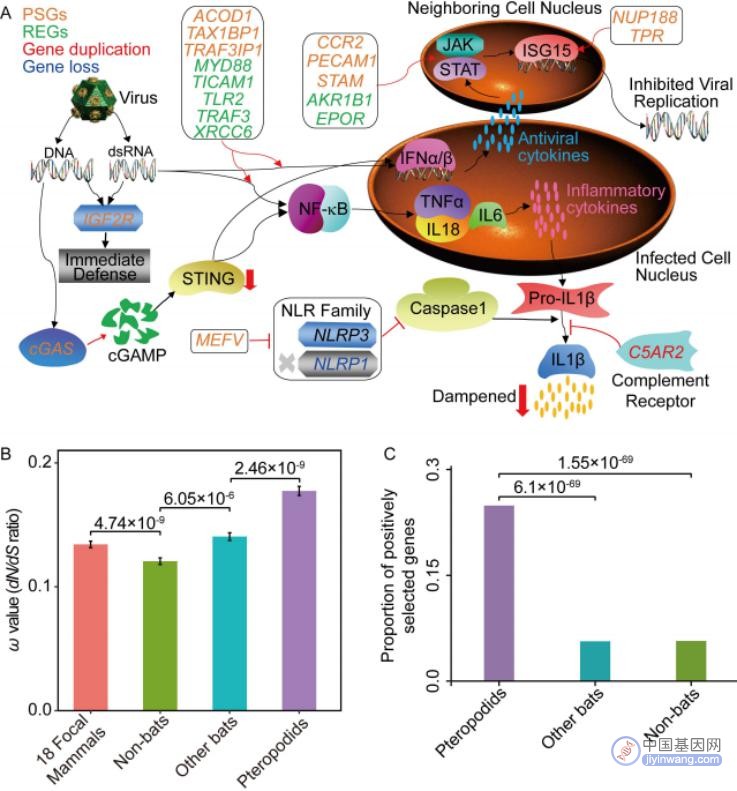
图5 狐蝠科特异性的免疫基因分子适应机制
MyD88突变抑制狐蝠科蝙蝠中TLR2依赖的炎症反应
尽管越来越多的研究报道了蝙蝠潜在的免疫分子适应机制,但很少有实验验证蝙蝠特异性变异对表型的影响。论文对上述2,162个直系同源基因进行分析,鉴定出628个基因蛋白中至少有1个氨基酸在狐蝠科蝙蝠和其他蝙蝠之间存在理化性质差异,并发现这些基因的平均ω值显著高于其他1,534个免疫基因。进一步的从中筛选受到正选择/快速进化的基因,并鉴定到了快速进化基因TLR2和MyD88。它们是TLR-MyD88信号传导通路中的重要分子。特别是,研究发现了狐蝠科蝙蝠相对于人类的4个特异性氨基酸突变残基属于TLR2-MyD88复合物中MyD88蛋白与TLR2蛋白的关键分子接口(P169、S170、A208和Q237),并且这4个氨基酸突变都导致MyD88蛋白的理化性质改变。
最后,论文针对狐蝠科MyD88的4个特异性突变设计了细胞分子实验(图6)。首先,通过在狐蝠Paki细胞进行免疫共沉淀(Co-IP)实验,证实了MyD88蛋白的特异性突变降低了其与Toll样受体TLR2的结合亲和力。接着,还分别在黑妖狐蝠Paki细胞和人类PEAKrapid细胞中进行了过表达实验,证明了MyD88蛋白的特异性突变减弱其对TLR2依赖性炎症因子的诱导。总之,这些结果揭示了蝙蝠在应对病毒感染时,具有系统性的免疫反应下调。
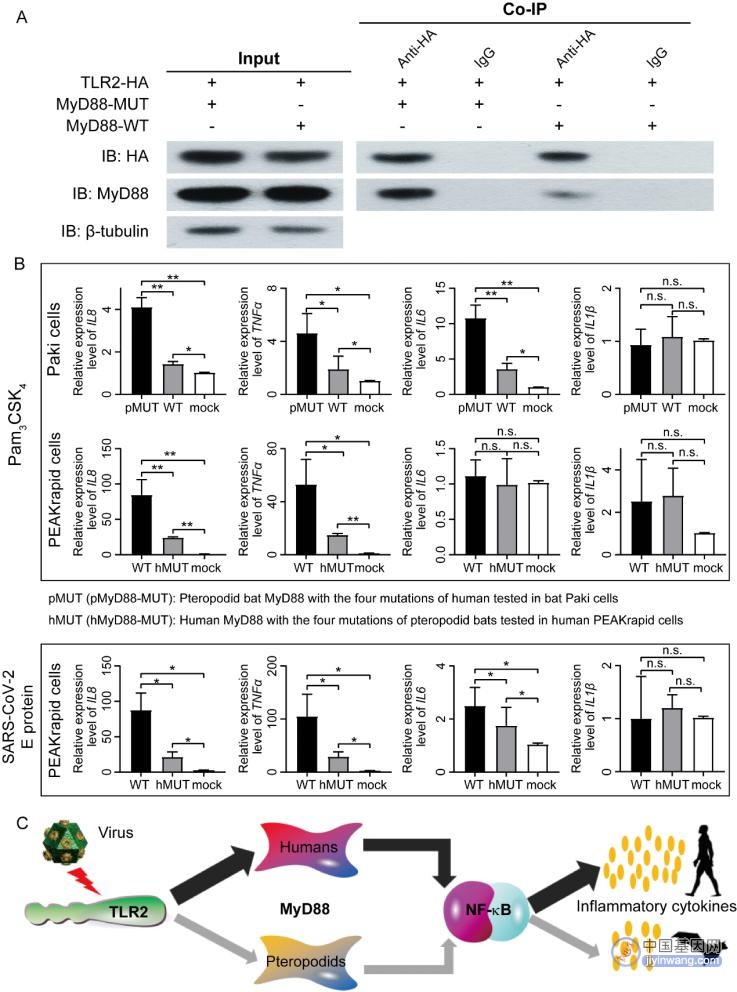
图6 狐蝠科蝙蝠MyD88特异性突变抑制炎症反应的分子实验证据
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。





")















