FDA与EMA对待细胞和基因疗法 (CGT) 申请的区别
01
术语辨析
首先需要指出的一点是,FDA 和 EMA对于细胞与基因疗法的术语设定是不一致的。细胞与基因疗法(CGT, cell and gene therapy)是FDA采用的称呼。对于同类治疗手段,EMA采纳的术语是“先进疗法药物产品”(ATMP, advanced therapy medicinal products)。不得不说,在命名的方面,还是FDA更合理、直接、明了一些;而EMA的称呼就有些笼统而且带有“歧视性”了,那些小分子、多肽、寡核苷酸和蛋白类的药物或许发问,“说谁不先进呢”?当然这只是句戏谑,大家记住各自的命名就好了。在EMA体系里,先进疗法药物产品(ATMP, advanced therapy medicinal products)之下,进一步涵盖了基因治疗药物 (GTMP,gene therapy medicinal product)、体细胞治疗药物 (SCTMP,somatic cell therapy medicinal product)、组织工程疗法 (TET,tissue-engineered therapies) 和联合先进疗法(combined advanced therapies)。为了论述方便,我们在文章中采用FDA的CGR称谓。
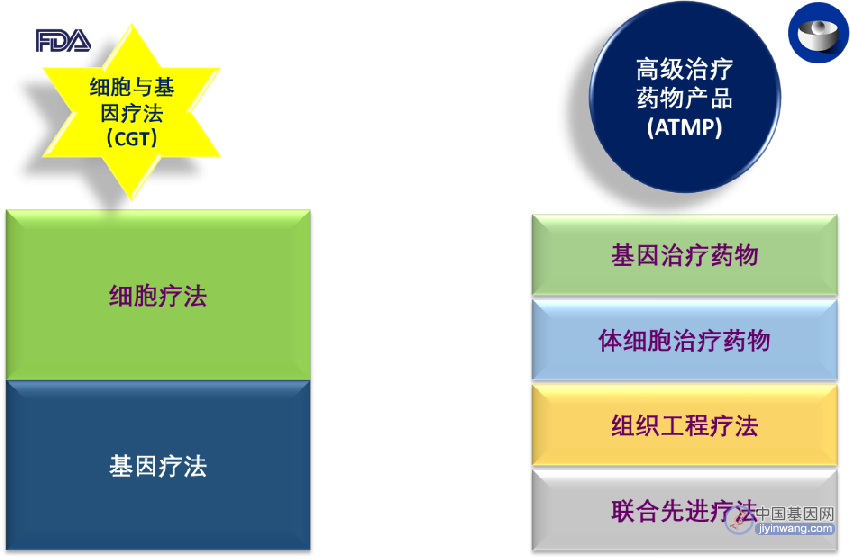
基因疗法与细胞疗法之间,也是有所区别但又相互联系的,毕竟基因来自于细胞内。细胞疗法旨在通过恢复或改变某些细胞组分,或使用细胞携带治疗剂在全身实现疾病治疗。 在细胞疗法中,细胞在注射到患者体内之前,在体外进行了培养或修饰。这些细胞可能来自患者(autologous cells,自体细胞)或供体(allogeneic cells同种异体细胞)。[3]
基因疗法旨在通过替换、灭活基因,或将基因引入细胞来治疗疾病,无论是在体内(in vivo)还是体外(ex vivo离体)。[2]
一些疗法被认为同时是细胞疗法和基因疗法。这些疗法通过改变特定类型细胞中的基因,并将其植入体内来发挥作用。
02
审批概况
迄今为止,FDA 已经批准了 27 种细胞与基因疗法(CGT),其中包括14 种细胞疗法和 13 种基因疗法。迄今为止,EMA 已经批准了 24 个 CGT:16 个 GTMP(基因治疗药物)、4 个 SCTMP (体细胞治疗药物)和 4 个 TET(组织工程疗法)。有 13 种产品获得了FDA和EMA的共同批准,包括已证明在治疗血癌方面非常有效的 CAR T 细胞疗法(Kymriah®、Yescarta®、Tecartus® 等)。
FDA 批准了八种脐带血产品(从脐带血中提取的干细胞)来治疗造血系统疾病,而 EMA 尚未批准任何一种产品,尽管有两种产品获得了孤儿指定:NiCord 和 NLA101。相反,Holoclar 在 EMA 被批准作为治疗角膜组织的干细胞疗法,而它在美国仅获得孤儿指定。在获得批准后,没有FDA批准的产品退出市场,而 24 种 EMA产品中有 7 种已经退市。
03
临床试验设计
在这两个地区,CGT发起人在证明安全性和有效性时面临着类似的挑战。对于目标适应症而言,患者人数通常很少,因为CGT针对的很多疾病都属于罕见病。对于那些危机生命,而又没有获批治疗的适应症来说,对征募参加临床试验的患者使用安慰剂的做法是不道德的。因此,不能采用黄金标准的随机对照试验 (RCT,randomized control trial)。但幸运的是,赞助商可以从孤儿药开发商那里借鉴策略,作为CGT临床设计的参考。
赞助商可以使用护理标准 (SOC,standard of care) 代替安慰剂组。在没有 SOC的情况下,自然历史研究(natural history study)或观察性研究,可以提供在没有治疗的情况下疾病进展的对比参照。但采用此策略可能会导致进行批准后研究作为获批条件。
随机对照试验的另一个标志是“经典”终点,它与所治疗的病症明显相关。比如无进展生存期(PFS, progression-free survival)和客观缓解率(ORR, objective response rate)。许多 CGT 开发人员依靠“替代”终点(可能预测长期临床获益的终点)来证明疗效。例如,血压可以预测心血管疾病的死亡率。如果替代终点经验证,可以显示与预期临床结果的相关性,则机构对这种方法持开放态度。
开发商和监管机构正在开发随机对照试验的替代方案,包括篮式设计(basket trials)、伞式设计(umbrella trials)和平台试验(platform trials)(统称为适应性临床试验adaptive clinical trials),它们可以结合真实世界的证据。
04
向FDA申请CGT
FDA的生物制品评估和研究中心 (CBER,Center for Biologics Evaluation and Research) 负责监管细胞治疗产品、人类基因治疗产品以及与细胞和基因治疗相关的设备。CBER 使用《公共卫生服务法》和《联邦食品药品和化妆品法》作为监督的授权法规。细胞治疗产品包括细胞免疫疗法、癌症疫苗,以及其他类型的,用于治疗某些适应症的自体和同种异体细胞,包括造血干细胞以及成人和胚胎干细胞。
美国细胞和基因治疗相关研发持续快速增长,多项产品在临床开发中取得进展。除了临床研究的监管监督外,CBER 还为新产品开发领域的医学研究人员和制造商提供积极的科学和监管建议。CGT的赞助商遵循与其他疗法相同的程序:提交 IND 申请以获得批准、进行临床试验、准备并提交生物制品许可申请 (BLA) 以供批准、里程碑会议(例如,pre-BLA 等),以及回答监管机构问题。发起人应在提交 IND 的同时请求加入再生医学高级治疗 (RMAT,Regenerative Medicine Advanced Therapy) 计划。RMAT 加速了有前途的疗法的开发,这些疗法可以治疗、修改、逆转或治愈严重的或危及生命的疾病,而这些疾病目前的医疗需求尚未得到满足。尽管申办者分别申请每个项目,但可以访问快速通道,例如快速通道指定(fast track designation)、突破性疗法指定(breakthrough therapy designation)、加速批准指定(accelerated approval)和优先审评指定(priority review designation)。RMAT 的其他优势包括:(1) 召开会议讨论替代终点以支持加速批准;(2) 就批准后要求(例如额外的临床试验)达成一致。
05
向EMA申请ATMP
所有先进疗法药物产品均通过欧洲药品管理局 (EMA) 集中授权。它们受益于单一的评估和授权程序。与所有药物一样,EMA在先进疗法药物产品获得批准和上市后继续监测其安全性和有效性。EMA还向开发商提供科学支持,帮助他们设计用于监测这些药物安全性的药物警戒和风险管理系统。
在提交ATMP申请的过程中,赞助商遵循与其他疗法产品相同的程序:提交和接收临床试验申请 (CTA,clinical trial applications) 的批准、进行临床试验、准备和提交上市许可申请 (MAA,marketing authorization application) 以供批准、请求科学建议,以及回答监管机构问题。负责监督 ATMP的 EMA机构是先进疗法委员会 (CAT,Committee for Advanced Therapeutics),而不是人用医药产品委员会 (CHMP,Committee for Medicinal Products for Human Use)。
符合条件的赞助商可以申请 EMA 的 PRIME 计划,以优先开发针对未满足需求的药物。赞助商可以收到额外的反馈(例如临床试验设计),并且该疗法有资格进行加速评估。EMA 会指定一个专门的联系人负责治疗的审查。此人甚至可以从卫生技术评估 (HTA) 机构获得反馈,以促进报销谈判。类似于FDA系统,可申请孤儿药指定。
参考文献:
[1] Cooper, M. How Do Cell & Gene Therapy Requirements Differ Between FDA & EMA? Cell&Gene. 24, 01, 2023.
[2] Friedman, T. A brief history of gene therapy. Nat Genet. 1992; 2: 93-98.
[3] American Society of Gene & Cell Therapy. Different approaches.
https://www.asgct.org/education/different-approaches
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。













全景分析:临床、上市、融资、市场规模")







