最全综述:淋巴瘤进行哪些基因检测可以辅助诊疗?
新的淋巴瘤分类(成熟淋巴肿瘤国际共识分类和第5版WHO淋巴肿瘤分类)将遗传学作为淋巴瘤诊断的一个组成部分,促使更好的淋巴瘤细分,患者风险分层和治疗反应预测。淋巴瘤存在一些高频疾病特异性突变,大多数亚型具有异质性分子图谱,突变基因很多。其中大多数发生频率较低,反映了淋巴瘤的临床异质性。多项研究识别了改善诊断和预后的分子标志物,NGS正成为临床实验室的重要工具。本综述为淋巴瘤提供了NGS指导。讨论了最常见的成熟淋巴瘤亚型中具有诊断、预后和预测潜力的基因变异,并提出了用于B细胞和NK/T细胞淋巴瘤突变和拷贝数变异检测的靶向测序panel。
研究背景
成熟淋巴瘤(霍奇金淋巴瘤(HL)和非霍奇金淋巴瘤(NHL))是最常见的血液系统实体肿瘤。随着高通量分子检测的开展,对淋巴瘤分子特征的了解加深。淋巴瘤的分类仍然主要基于形态学、免疫表型和一些遗传特征。成熟淋巴瘤的新分类,成熟淋巴肿瘤国际共识分类(ICC)和第5版世界卫生组织(WHO)淋巴肿瘤分类,已经纳入了新开发的技术来改善淋巴瘤分类,分子变异现在是淋巴瘤诊断标准的一部分。
正在使这些高通量技术标准化,以实现其全面和成功的临床应用,但关于测序方法仍然存在很多争议。最关键的问题之一是测序panel的选择和组成。理想的panel应有助于诊断、预后、治疗选择和监测,但要足够小,以便广泛和统一使用。对整个外显子组或基因组进行测序的能力正在稳步增长。尽管如此,定制panel是目前扩大此方法适用性的最可及的选择。这些靶向panel能够以较高的灵敏度和较低的成本深入地分析少量基因。可以使用基于扩增子或捕获的测序panel。基于捕获的panel不仅可以检测单核苷酸变异(SNV)和插入缺失(indel),还可以检测拷贝数变异(CNA)和一些结构变异。然而,结构变异的检测需要进一步改善。此外,样本管理、panel组成、测序程序、生物信息学分析和变异解释的标准化对于开发有用的临床工具至关重要。
本综述旨在总结各种淋巴瘤亚型的重要分子特征,描述与每种亚型相关的基因,大规模测序或NGS panel可包含这些基因。
成熟B细胞淋巴瘤
慢性淋巴细胞白血病
慢性淋巴细胞白血病(CLL)是一种低级别淋巴组织增生性疾病,其特征是血液、骨髓、淋巴结和脾脏中成熟(通常CD5+)B细胞克隆性增殖和积蓄。
超过80%的CLL患者存在一些细胞遗传学异常,根据荧光原位杂交(FISH)检测结果将患者分为不同的风险组:约55%的患者发生13号染色体长臂缺失(del(13q));12号染色体三体是第二常见的染色体异常(发生率为10%-20%);约25%的未接受过化疗的晚期患者和10%的早期疾病患者伴有11号染色体长臂缺失(del(11q));5-8%的未接受过化疗的患者伴有17号染色体短臂缺失(del(17p))。只有del(17p)被认为是显著不良预后因素。CLL中其他常见的异常包括6q缺失(5%)和2p扩增(5-16%)等。
免疫球蛋白重链可变区(IGHV)体细胞超突变(SHM;IGHV序列相似性<98%,突变的CLL,M-CLL)的预后优于没有突变(未突变的CLL,U-CLL)。最近发现,在CLL中发生率约为5-15%的IGLV3-21R110突变可导致预后不良,与IGHV突变状态无关。TP53突变见于4-37%的CLL患者,可以单独发生,也可以与del(17p)共存,后者更常见。TP53突变与对化疗敏感性较低和总生存期(OS)较短有关。NGS研究识别了其他与预后相关的基因突变,如BIRC3,NOTCH1,SF3B1,MYD88,ATM,FBXW7,POT1,NFKBIE,CHD2,RPS15,IKZF3,ZNF292,ZMYM3,ARID1A和PTPN11。
Richter转化定义为CLL转化为侵袭性淋巴瘤,最常见的是弥漫性大B细胞淋巴瘤(DLBCL)。这些患者对传统化疗的反应通常比新发DLBCL差。此外,其突变模式与DLBLC,非特指型(NOS)不同。Richter转化的风险与既往治疗、U-CLL、NOTCH1突变、del(17p)和del(11q)有关。
BTK、PLCG2和CARD11突变与BTK抑制剂耐药有关,BCL2突变与维奈克拉耐药有关。因此,CLL在免疫化疗和靶向治疗之间的选择很大程度上取决于17p/TP53和IGHV状态。尽管治疗选择并不严格要求测序数据,但这些数据应纳入决策和随访。如上所述,一些分子变异是预后不良因素,似乎导致对常规化疗的反应较差,在考虑基于BTK或BCL2抑制剂的其他治疗方案时可能会有所帮助。
理想的用于CLL的NGS panel应包括IGHV SHM,基因突变和CNA检测(表1和图1)。
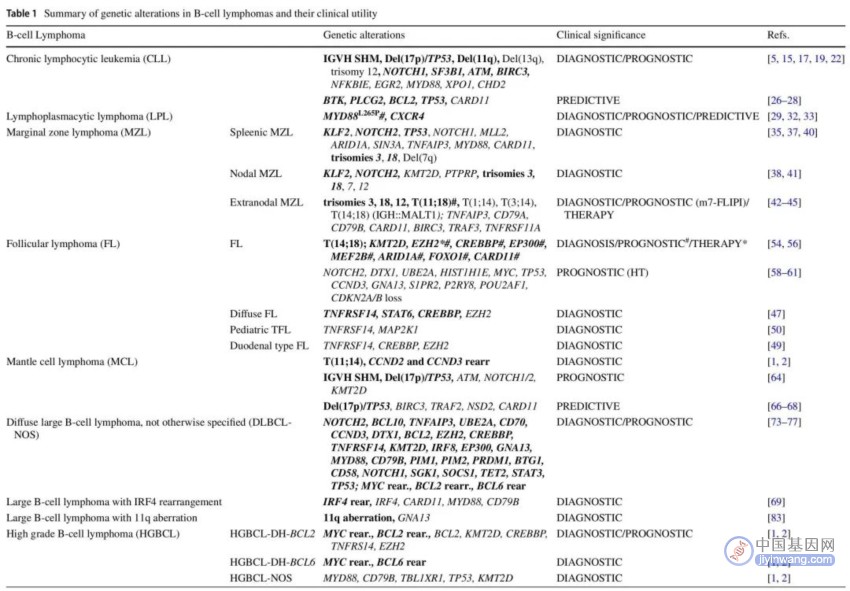

表1. B细胞淋巴瘤中的基因变异及其临床意义
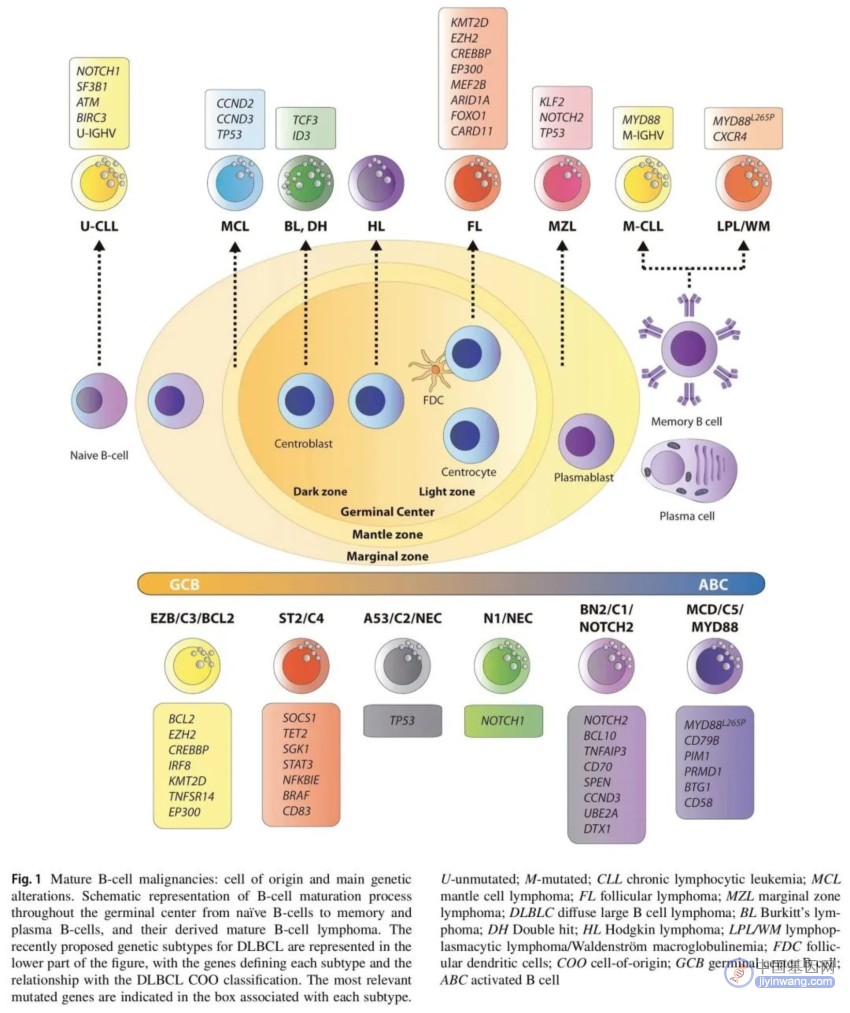
图1. 成熟B细胞恶性肿瘤的细胞起源和主要基因变异
淋巴浆细胞淋巴瘤/华氏巨球蛋白血症
淋巴浆细胞淋巴瘤/华氏巨球蛋白血症(LPL/WM)是一种罕见的低级别B细胞淋巴瘤,年发病率为3-4例/百万人,特征为克隆性淋巴浆细胞骨髓浸润和免疫球蛋白M增高。由于缺乏特异性形态学、免疫表型或染色体特征,需要在排除其他小B细胞淋巴瘤后做出诊断。
尽管罕见,但近年来,随着MYD88和CXCR4基因高频突变的鉴定,我们对该疾病生物学的了解显著改善。超过90%的LPL/MW患者携带MYD88L265P突变。不过,这对于诊断非必要,不具有特异性,可见于其他B细胞淋巴瘤,例如非生发中心型(GC)DLBCL、CLL、脾边缘区淋巴瘤、原发性皮肤DLBCL、腿型DLBCL、原发性中枢神经系统DLBCL或睾丸DLBCL。然而,两种突变同时出现高度提示LPL/WM。MYD88L265P通过BTK触发肿瘤细胞生长,BTK是伊布替尼的靶标。CXCR4突变见于30%的LPL/WM患者,与较短的无治疗生存期相关,并导致对伊布替尼的耐药性。MYD88和CXCR4突变患者的疾病表现和对伊布替尼治疗的反应存在显著差异:有MYD88L265P、无CXCR4突变患者的总体缓解率为100%,共突变患者为85.7%,无突变患者为71.4%。
其他分子变异包括6q缺失(40%-60%的患者)、PRDM2和BTG1(约90%的患者)、HIVEP2、MKLN1、PLEKHG1、LYN、ARID1B、FOXP1和ARID1A突变。分子突变诊断方法,特别是NGS,有助于完善LPL/WM的诊断标准。除了辅助诊断,分子检测还可用于LPL/WM的风险分层和治疗计划(表1和图1)。
边缘区淋巴瘤
边缘区淋巴瘤(MZL)是一组起源于边缘区B淋巴细胞的惰性B细胞淋巴瘤。主要为脾MZL(SMZL),伴或不伴绒毛淋巴细胞、淋巴结MZL(NMZL)和黏膜相关淋巴组织结外MZL(EMZL或MALT)。这些是不同的临床亚型,具有特定的诊断标准和不同的遗传特征、临床行为和治疗意义。MZL约占淋巴结边缘区GC后记忆B细胞起源的所有NHL的10%。有研究探索了MZL的基因图谱,揭示了不同亚型的突变基因存在相当大的重叠(表1和图1)。
SMZL中最广泛突变的基因是KLF2和NOTCH2。TP53和NOTCH1突变也较为常见。另外还有导致NF-κB通路激活的TNFAIP3、CARD11、MYD88(p.265热点外)或TRAF3突变,以及KMT2D、ARID1A和SIN3A等染色质重塑基因突变。超过30%的脾MZL伴有7q31-32缺失。
NMZL和SMZL有几个共同的变异。3号和18号染色体三体见于约 25%的SMZL和NMZL患者。NMZL的突变谱显示其他常见克隆异常,如7和12三体以及6q缺失。突变分析识别了KLF2,PTPRD,KMT2D,NOTCH2,LRP1B,TET2和TNFRSF14突变。NMZL中其他相对少见的分子变异包括BRAF,EZH2和HIST1H1E突变。
EMZL/MALT的发病机制与几种高频染色体异常有关,例如3、12、18三体,见于20-30%的患者。EMZL中几种染色体易位也较常见。最常见的是t(11;18)(p21;q21),导致BIRC3和MALT1之间产生功能性嵌合融合产物。该易位与对抗生素的反应率低、幽门螺杆菌阴性、更晚期的疾病以及转化为DLBCL的风险较低有关。该易位对 EMZL具有特异性,在SMZL或NMZL中尚未报告。其他常见的易位包括t(3;14)(p14;q32)(FOXP1::IGH),见于约10%的EMZL患者;t(14;18)(q32;q21)(IGH::MALT1),存在于15-20%的非胃肠道EMZL中;t(1;14)(p22;q32)(BCL10::IGH)是一种罕见的易位,见于1-2%的EMZL。EMZL中有TNFAIP3(A20)所在染色体区域6q23纯合缺失的报道,可能通过诱导组成型NF-κB活化来促进淋巴瘤发生。MYD88突变可见于眼附属器MALT淋巴瘤(5%的患者),可激活NF-κB、STAT3和AP1转录因子。除了TNFAIP3和MYD88突变外,还发现了其他NF-κB调节因子变异(CD79A、CD79B、CARD11、BIRC3、TRAF3和TNFRSF11A)。上述参与NF-κB通路激活的高频分子变异是MALT淋巴瘤的潜在治疗靶点。
滤泡性淋巴瘤
滤泡性淋巴瘤(FL)是一种起源于GC B细胞的恶性肿瘤,是最常见的惰性B细胞淋巴瘤。FL仍然是一种无法治愈的恶性肿瘤,但OS可能达20年。FL的一个关键标志是t(14;18)(q32;q21)IGH::BCL2易位,这是其肿瘤发生过程中的第一次打击。最近,新的WHO和ICC分类列出了独特的FL亚型,如原位滤泡性B细胞肿瘤,十二指肠型FL,原发性皮肤滤泡细胞淋巴瘤,儿童型FL和睾丸FL。
2009年首次描述了一种没有t(14;18)的独特弥漫性滤泡性淋巴瘤(dFL)亚型。在最近的一项研究中,NGS分析确定了两个分子集群:一个集群的特征为TNFRSF14突变;另一个集群分子变异较少,但注意到一个亚群具有STAT6和CREBBP共突变,没有TNFRSF14和EZH2突变。这些发现提示dFL可能为一种t(14;18)阴性FL亚型。
胃肠道FL,尤其是十二指肠型FL(DTFL),常为结外FL。这种淋巴瘤常见于十二指肠的第二部,表现出惰性的临床行为。在形态学和免疫表型上与典型的FL难以区分。NGS的更广泛应用发现,TNFRSF14、CREBBP和EZH2等高频突变基因的突变频率与典型FL没有显著差异,但KMT2D的突变频率在DTFL中较低。
儿童型FL(PTFL)发生于年轻患者,好发于头颈部。一些研究使用NGS技术描述了PTFL的特定突变谱,与其他淋巴瘤(包括典型FL)不同。TNFRSF14和MAP2K1是PTFL中最常报告突变的基因。约80%的患者存在TNFRSF14或MAP2K1突变,但这两个基因突变通常不共存。这一发现提示,这两个基因对PTFL的发生至关重要。
2011年,Morin等人描述了KMT2D和其他染色质修饰基因(CMG)高频突变。这些基因编码组蛋白甲基转移酶(EZH2,KMT2D)或组蛋白乙酰化酶(MEF2B,CREBBP,EP300)。这些基因变异已被确立为FL的核心分子标志,对于确定GC和GC后B细胞的命运至关重要。FL与其他B细胞淋巴瘤的区别特征是CMG突变率高。FL中突变率最高的CMG是KMT2D(72%),其次是CREBBP(约占FL的65%)和EP300(15%)。在25%的FL中发现EZH2突变,与预后相关,目前正探索其作为具有治疗潜力的药物靶点。EZH2是GC表型的调节因子,该基因突变阻断B细胞并阻止其分化为浆细胞。
靶向表观遗传失调是一个有吸引力的概念。然而,最常见的CMG突变(KMT2D和CREBBP)是功能丧失/蛋白质丧失事件,很难用药物靶向。这种限制促使研究人员关注EZH2突变,多个公司都在开发EZH2抑制剂。在最近的一项II期研究中,口服EZH2抑制剂tazemetostat在复发或难治性FL患者中显示出抗肿瘤活性。有或没有EZH2突变的患者接受tazemetostat治疗,69%的EZH2突变患者和35%的EZH2野生型患者获得客观缓解。
滤泡性淋巴瘤国际预后指数(FLIPI)是使用最广泛的风险预测指标。关于无失败生存,Pastore等人提出了一种临床基因风险模型,即m7-FLIPI评分,包括7个基因(ARID1A,EZH2,EP300,FOXO1,MEF2B,CREBBP和CARD11)的突变状态,东部肿瘤协作组(ECOG)体能状态和FLIPI。m7-FLIPI在接受一线利妥昔单抗联合化疗(CVP或CHOP)的患者中界定了一个高风险组,其5年无失败生存率为38%,而低风险组为77%。但低风险m7-FLIPI并不表明病程更惰性,因为所有患者都需要化疗。然而,几项研究得出结论,m7-FLIPI临床基因模型的预后价值似乎取决于治疗方案(表1和图1)。
研究显示,组织学转化(HT)发生于15-30%的FL患者。HT是指FL 演变为临床侵袭性淋巴瘤,如DLBCL或伯基特淋巴瘤,通常与预后不良和化疗耐药性有关。HT与使细胞周期进程和DNA损伤反应失调的变异(CDKN2A/B、MYC和TP53),或其他在转化样本中发生率高于FL肿瘤的基因变异(CCND3、GNA13、S1PR2和P2RY8)有关。一项研究分析了转化或未转化患者的FL样本,发现诊断时FL活检样本存在四个基因(NOTCH2、DTX1、UBE2A和HIST1H1E)突变与转化时间较短有关。该研究还识别了转化时富集的突变基因,如POU2AF1,其在GC结构和迁移中起作用。
套细胞淋巴瘤
套细胞淋巴瘤(MCL)约占NHL的6%。该疾病有两种临床表现:常见的经典MCL(cMCL)(90%的患者)通常具有侵袭性临床病程(SOX-11阳性细胞和IGHV未突变),以及白血病样非淋巴结性MCL(nnMCL)通常具有惰性临床表现(10%的患者)(SOX-11阴性、CCND1和TLR2突变以及IGHV体细胞超突变)。MCL通常是一种侵袭性和无法治愈的B细胞恶性肿瘤,但一些患者可能具有惰性的临床病程。
MCL的特征是t(11;14)(q13;q32)易位,导致细胞周期蛋白D1(CCND1)过表达,在近95%的患者中检测到。在不明成熟B细胞肿瘤中检测CCND1有助于支持MCL诊断,FISH是目前用于识别高频细胞遗传学变异的金标准检测方法,尽管这种方法可能无法检测到复杂或隐性重排。少数没有该特征性CCDN1重排的患者具有CCND2或CCND3易位的特征。
一些研究识别了显著突变的基因,如ATM和肿瘤抑制因子TP53,在预后较差的侵袭性肿瘤中发现了NOTCH2突变。
MIPI(MCL国际预后指数)基于体能状态、年龄、乳酸脱氢酶(LDH) 水平和白细胞计数的加权总和。正在探索其他修改,如“MIPI遗传”(MIPIg)以完善该分数。当存在KMT2D突变和TP53缺失或突变时,MIPIg与进展和死亡风险增加有关。在诊断时,TP53突变频率约为11-25%,但在复发时增加到45%。TP53缺失(由FISH测定)和TP53突变与最差生存率相关(表1和图1)。
伊布替尼难治性MCL患者生存率低,缺乏最佳治疗策略。一些研究探索了BIRC3、TRAF2和CARD11基因突变,MCL进展和伊布替尼耐药之间的关系。最近,一项研究分析了伊布替尼治疗后出现疾病进展或疾病转化的患者亚群的突变谱。使用靶向NGS,在75%的伊布替尼进展患者中检测到TP53变异。观察到染色质修饰基因突变,如75%的经伊布替尼治疗发生转化的MCL患者检测到NSD2突变。得出结论,NSD2突变与改变的甲基化和染色质功能障碍有关,导致基因表达异常,在MCL进展和伊布替尼耐药中具有病理意义。
弥漫性大B细胞淋巴瘤
DLBCL是最常见的NHL亚型(30%-35%的患者),其特征是大的成熟B细胞形态和表型。DLBCL在临床行为及病理和分子诊断方面具有异质性。最常见的DLBCL类型(80%)是DLBCL-NOS,没有特定的临床表现或病理。基于起源细胞(COO),WHO分类识别了三种分子亚型:生发中心(GCB),活化B细胞(ABC)或非GCB,以及不可分类的DLBCL。这种分类具有预后价值,ABC-DLBCL与较差的结局相关。然而,COO并不意味着不同的治疗方案,所有亚型的治疗均以R-CHOP(利妥昔单抗、环磷酰胺、阿霉素、长春新碱和泼尼松龙)为主。高达40%的DLBCL对一线治疗不敏感或复发。
一些研究人员最近提出了新分子亚群,具有广泛一致性,提示突变分析可能是对DLCBL进行分类的有希望的替代方案,具有预后和诊疗一体化价值。每个分子集群都有独特的突变谱。Schmitz等人界定了以下亚型:MCD(MYD88L265P和CD79B共突变),BN2(BCL6融合和NOTCH2突变),N1(NOTCH1突变),EZB(EZH2突变和BCL2易位),A53(TP53突变和缺失)和ST2(SGK1和TET2突变)。另一种方法区分了五个DLBCL亚群,包括两个ABC-DLBCL组,一个具有低风险和可能的边缘区起源(C1),另一个是高风险组(C5),在MYD88,CD79B和PIM1突变患者中富集;结局较好(C4)和较差(C3)的GC-DLBCL亚群;以及TP53双等位基因失活、CDKN2A缺失和相关基因组不稳定的与ABC/GC无关的亚群(C2)。为了统一这两种分类,Lacy等人根据每个亚型中富集度最高的突变基因确立了NOTCH2,MYD88,BCL2,TET2 / SGK1和SOCS1 / SGK1亚型。然而,所有这些研究主要基于全基因组测序,不适用于常规临床实践。因此,有研究提出了简单可行的分类,结合了代表每个遗传亚型的一组选定基因的突变和易位数据。
可能很快就会达成共识,开发一个用于分子分类的NGS panel(表1和2,图1)。为了实现个体化医疗,至少应包括对MYC,BCL2和BCL6重排的分析,诊断时的COO确定以及选择的NGS panel。虽然关于分子亚型的分类没有达成一致意见,但可以设计一个充分的初步NGS panel,包括一组来自分子分类的常见基因,MCD亚型:MYD88,CD79B,PIM1,PIM2,PRDM1,BTG1,CD58,ETV6和TBL1XR1;BN2亚型:NOTCH2,TNFAIP3,BCL10,UBE2A,CD70,CCND3和DTX1;EZB亚型:EZH2,CREBBP,TNFRSF14,KMT2D,BCL2,IRF8和EP300;ST2亚型:SGK1、SOCS1、TET2、NFKBIA和STAT3;N1亚型:NOTCH1。

表2. DLBCL分子亚型
起源于免疫赦免部位的结外DLBCL,例如原发性中枢神经系统DLBCL(PCNSL),原发性睾丸DLBCL,原发性皮肤DLBCL,腿型和其他相关亚型,如乳腺DLBCL,具有相似的分子特征,最近对其分类存在一些争议。大多数这些部位的淋巴瘤是非GCB/ABC型,似乎具有共同的分子特征,如MYD88L265P和CD79B突变率高,这是DLBCL-MCD/C5/MYD88分子亚型的特征。
与病毒药物(EBV相关、HHV-8相关)相关的DLBCL很少见,在大多数情况下,其诊断基于临床表现和病理特征。在EBV阳性DLBCL中描述了一组可能对该疾病具有特异性的突变基因,包括CCR6、CCR7、DAPK1、TNFRSF21、CSNK2B和YY1,以及常见的PD-L1分子变异。
伴有IRF4重排的大B细胞淋巴瘤常见于年轻患者,好发于咽淋巴环和颈部淋巴结。一项针对儿童LBCL的研究发现,伴有IRF4重排的亚型具有GCB表型,IRF4和NF-κB通路基因(CARD11,MYD88,CD79B)突变频率高。
伴有11q异常的LBCL(在第5版WHO分类中命名为“伴有11q异常的HGBCL”)在之前的WHO分类中被归类为伴有11q异常的伯基特样淋巴瘤。突变模式更像GCB-DLBCL而不是伯基特样,缺乏MYC重排,GNA13突变频率高。ID3-TCF3复合体基因变异在LBCL-11q中少见。
高级别B细胞淋巴瘤
最近修订的第5版WHO造血与淋巴组织肿瘤分类和ICC目前确定了以下亚群:伴有MYC和BCL2重排的高级别B细胞淋巴瘤(HGBCL)(伴或不伴BCL6重排,HGBCL-DH-BCL2),伴有MYC和BCL6重排的暂定亚型(HGBCL-DH-BCL6)和HGBCL-NOS。
HGBCL-DH-BCL2淋巴瘤亚型的突变相对同质,与FL相似,常见BCL2,KMT2D,CREBBP,TNFRS14和EZH2分子异常。最近的几项研究描述了DLBCL患者的基因表达特征(GEP)与HGBCL(DH样GEP特征)相似,MYC、BCL2、DDX3X、TP53和KMT2D突变率较高(表1和图1)。
具有双重MYC和BCL6重排的淋巴瘤现在被认为是一种DLBCLNOS或HGBCL亚型,其基因表达谱具有高度异质性,与伴有MYC和BCL2重排的DLBCL/HGB细胞淋巴瘤无关。
HGBCL-NOS被认为是另一种亚型,涵盖其他亚型未包括的患者。从分子角度来看,这是一个异质性类别,包括伴有MYD88,CD79B或TBL1XR1突变的活化B细胞淋巴瘤。TP53和KMT2D突变最常见。基因表达谱显示,大约一半的HGBCL-NOS患者具有先前描述的DH样GEP特征。
伯基特淋巴瘤
伯基特淋巴瘤(BL)是一种高度侵袭性成熟B细胞NHL,其特征是快速增殖。BL占成人淋巴瘤的1%-2%,是常见的儿童肿瘤。
过去描述了三种BL临床亚型目前被WHO分类认可:地方性,非地方性或散发性,以及免疫缺陷相关性。三种亚型表现出相同的形态学和免疫表型特征,但存在临床和流行病学差异。所有临床亚型具有共同的病理特征。BL的一个决定性特征是MYC组成性过表达,这是由于该癌基因与位于14号、2号和22号染色体上的三个免疫球蛋白基因之一的易位引起的,分别占患者的近80%、15%和5%。其他典型的淋巴瘤易位,如BCL2和BCL6,未在BL中观察到。然而,MYC失调不足以导致淋巴瘤发生。因此,通常使用NGS检测其他分子变异。
将BL与其他HGBCL区分开来是临床实践中必须解决的重要挑战。基因表达谱研究揭示了独特的BL特征,使其成为一个独立的亚群。几项研究探索了BL分子图谱,表明ID3/TCF3依赖性中心母细胞基因表达程序,强直PI3K-AKT-mTOR信号传导以及细胞周期失调和凋亡是BL淋巴瘤发生的重要机制。肿瘤抑制基因(如TP53、CDKN2A和DDX3X)失活突变在BL中常见。此外,据报道,TP53失活是潜在的预后生物标志物,因为这些突变在难治性患者中富集。同样,男性中BL发生率较高的原因之一可能是DDX3X失活变异。DDX3X功能缺失突变缓和MYC驱动的全局蛋白合成,已建立的恶性细胞通过Y染色体同系物(DDX3Y)的异常表达恢复完整的蛋白质合成能力,DDX3Y表达通常仅限于睾丸。细胞迁移和播散障碍的原因之一是P2RY8和GNA13失活,高水平的增殖不仅是由于CCND3活化,还由于CDKN2A失活。另一种致癌机制是转录因子TCF3及其负调节因子ID3的突变导致BCR信号通路的组成性激活。
BCR信号通路激活PI3K-AKT-mTOR信号通路,突出靶向PTEN和FOXO1变异,对BL生存至关重要。最后,BL中常见各种染色质调节因子突变,很少观察到EZH2、CREBBP和KMT2D突变,与GCB-DLBCL不同。此外,寻找具有临床意义的新的BL细分策略的研究表明,EBV状态可能是一种新兴的方法。EBV阳性BL异常SHM水平显著较高,驱动突变较少,特别是在细胞凋亡通路,TCF3和ID3突变较少(表1和图1)。
新的WHO分类推荐将EBV阳性和EBV阴性BL视为不同的亚型。关于鉴别诊断,必须识别用于BL诊断的MYC易位,以及在伯基特样淋巴瘤中观察到的典型染色体 11q扩增/缺失模式。至于NGS,推荐分析TCF3和ID3(在BL中高频突变)以及EZH2、CREBBP和KMT2D(在其他淋巴瘤中突变)的差异突变模式。此外,TP53突变状态可提供预后信息。
霍奇金淋巴瘤
大多数霍奇金淋巴瘤(HL)是起源于B细胞的克隆性肿瘤。HL占所有淋巴瘤的25-30%,分为经典型霍奇金淋巴瘤(cHL)(95%的患者)和结节性淋巴细胞为主型霍奇金淋巴瘤(NLPHL)(5%的患者)。与其他肿瘤不同,肿瘤细胞(霍奇金和R-S细胞[HRS]以及淋巴细胞和组织细胞[L&H],在NLHPLN中)占总细胞数的不到1-2%。ICC和新WHO分类提出“结节性淋巴细胞为主型B细胞淋巴瘤”(NLPBL)作为NLPHL的新术语,基于其与cHL的显著生物学和临床差异以及与富于T细胞/组织细胞的大B细胞淋巴瘤的密切相关性。
cHL、原发性纵隔大B细胞淋巴瘤和纵隔灰区淋巴瘤是具有共同分子变异、表型和临床特征(包括前纵隔受累)的相关疾病。cHL是HRS细胞(及其变体)的单克隆增殖,伴有反应性微环境,识别这两种元素对其诊断至关重要。在细胞遗传学方面,除了非整倍体和超四倍体外,HRS细胞还表现出高频染色体失衡,包括2p13(REL),9p24.1(CD274(PDL1),PDCD1LG2(PDL2),JAK2),17q21(MAP3K14)扩增和6q23-q24(TNFAIP3)缺失。
肿瘤细胞的缺乏阻碍了cHL的遗传分析。然而,基于组织显微解剖或流式细胞术细胞分离的遗传研究表明了特定通路失调而不是特定基因突变。研究揭示了NF-κB通路成分(TNFAIP3,NFKBIA,NFKBIA,REL),JAK/STAT通路(SOCS1, PTPN1, STAT6, STAT3, CSF2RB),表观遗传调节因子如EP300、CREBBP和TP53,以及有利于逃避细胞凋亡和细胞增殖的免疫逃逸调节因子如MHC 1类成分B2M,MHC 2类反式激活因子(C2TA)基因和FAS基因失活突变这些高频体细胞突变。其他与cHL发生相关的信号通路包括MAPK/ERK、AP1、PI3K/AKT和NOTCH(表1和图1)。
成熟T细胞和NK细胞淋巴瘤
非霍奇金T细胞淋巴瘤(NHL-T)是一组相对罕见的异质性恶性肿瘤,起源于T淋巴细胞或自然杀伤(NK)细胞,通常具有侵袭性临床行为。在西方国家,T细胞淋巴瘤(TCL)占所有NHL的5%-10%,总体发病率为每年0.5-2.0/10万居民。根据组织学亚型,NHL-T首次出现时具有淋巴结或结外表现。最近的WHO和ICC分类将以下亚型确认为原发性淋巴结T/NK细胞起源的肿瘤:滤泡辅助性TCL(TFH),间变性大细胞淋巴瘤(ALCL;ALK阳性和ALK阴性),外周TCL非特指型(PTCL-NOS)以及原发性EBV阳性T-/NK细胞淋巴瘤。关于结外亚型,最常见的结外TCL亚型是皮肤TCL(CTCL)、结外NK/T细胞淋巴瘤鼻型(ENKL)、乳腺植入物相关间性变性大细胞淋巴瘤(BIA-ALCL)、肠TCL(ITCL)和肝脾TCL(HSTCL)(表3和图2)。
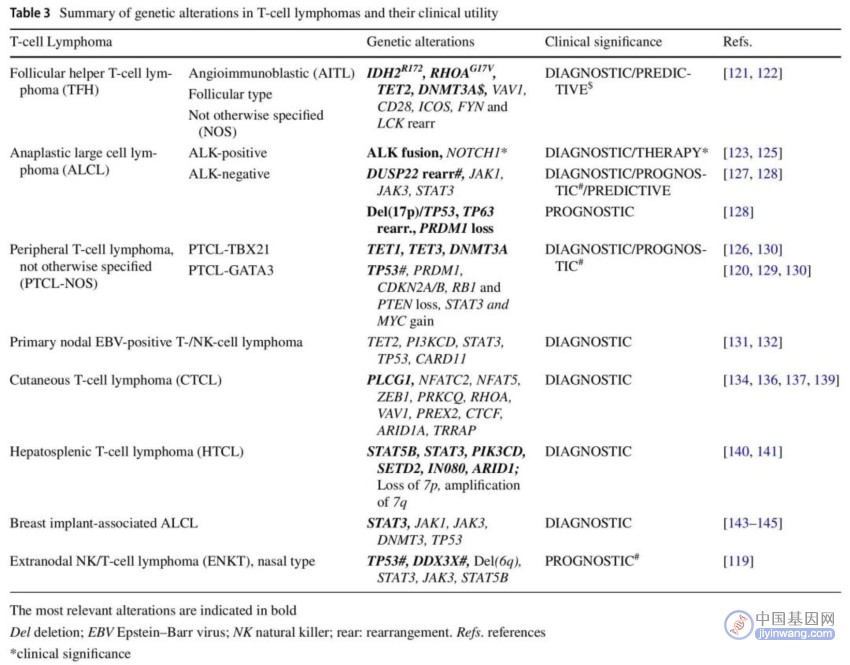
表3. T细胞淋巴瘤中的基因变异及其临床意义
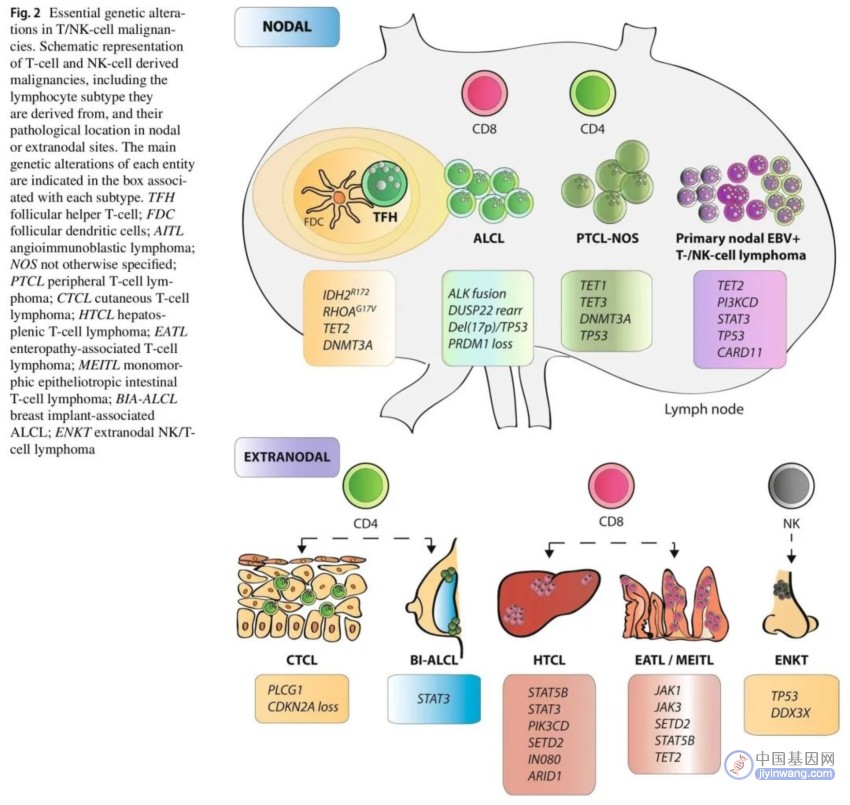
图2. T/NK细胞恶性肿瘤中的重要基因变异
淋巴结T/NK细胞淋巴瘤
滤泡辅助T细胞淋巴瘤
滤泡辅助T细胞(TFH)淋巴瘤包括三个具有共同分子特征和类似于定义该亚型的TFH细胞的基因表达特征的亚型:血管免疫母细胞型(AITL)、滤泡型和NOS。这些淋巴瘤具有相似的突变图谱,包括甲基化相关基因TET2(约80%的患者)和DNMT3A(30-40%)功能缺失突变。其他高频突变基因有CD28,RHOA(G17V)和IDH2(R172),主要见于一部分AITL,以及TCR信号通路基因。ICOS::CD28,ITK::SYK和涉及VAV1的融合是常见融合。总之,TFH淋巴瘤存在共同的表观遗传学和TCR信号通路基因变异。
间变性大细胞淋巴瘤
间变性大细胞淋巴瘤(ALCL)的特征是多形性肿瘤细胞表达CD30,包括四种不同的亚型:两种淋巴结亚型和两种结外亚型。本节重点介绍ALK阳性(ALK重排,几种不同的融合伴侣)和ALK阴性ALCL。最常见的ALK重排是 t(2;5)(p23;q35),其导致NPM1与ALK融合,产生嵌合蛋白。这些患者具有高频NOTCH1突变,可能为潜在的治疗靶点。TP53和表观遗传调节因子(EP300、KMT2D/C)突变也有报道。需要免疫表型、组织学、分子和临床数据来正确诊断ALK阳性ALCL。
ALK阴性ALCL是一种相当异质的亚型。DUSP22重排存在于约20-30%的患者,已被定义为一种独特的ALK阴性亚型。大约60%的ALK阴性患者JAK-STAT3激活,主要由JAK1、JAK3和STAT3突变或TYK2、ROS1和FRK重排导致。这种激活在DUSP22重排的ALK阴性ALCL中不存在。一小部分ALK阴性ALCL患者表现为TP63重排以及TP53和PRDM1缺失,与侵袭性病程有关。
外周T细胞淋巴瘤,NOS
外周T细胞淋巴瘤,NOS(PTCL-NOS)占淋巴结PTCL的34%,包括不符合纳入其他亚型纳入标准的患者。据报道,FAT1突变在PTCL-NOS中较常见,与预后较差相关。该类别分为两个分子亚组,PTCL-TBX21和PTCL-GATA3,两组具有不同的分子图谱。特征是死亡率高,预后差。PTCL-GATA3的分子图谱更复杂,预后更差。具有高频TP53和PRDM1缺失和突变,CDKN2A/B,RB1和PTEN缺失以及STAT3和MYC扩增。PTCL-TBX21表现出高频CMG突变,如TET1,TET3和DNMT3A。
原发性淋巴结EBV阳性T/NK细胞淋巴瘤
原发性EBV阳性T/NK细胞淋巴瘤在ICC和WHO分类中被标记为新的暂定亚型。该疾病较为罕见,发生于老年或免疫功能低下患者,预后不良。该亚型表现出低基因组不稳定性,EBV microRNA下调,以及STAT3,TET2,CARD11,BCOR,ARID1B,TP53和PI3KCD基因高频突变。
结外T/NK细胞淋巴瘤
皮肤T细胞淋巴瘤
皮肤T细胞淋巴瘤(CTCL)是起源于皮肤的异位淋巴组织增生性疾病的一种亚型。最常见的临床表现是蕈样肉芽肿(MF),患者表现为皮肤斑块、斑块或肿瘤。在组织学上,观察到CD4 +淋巴细胞浸润。塞扎里综合征(SS)是MF的一种白血病形式。SS和MF的全基因组,外显子组和靶向测序揭示了基因变异,包括某些基因体细胞突变和CNA,这些基因基因参与已知的关键细胞活动,如DNA损伤(如TP53(mut和del)和ATM(主要是del)),TCR信号通路(PLCG1,ZEB1(del)),NF-κB信号通路(CARD11和TNFRSF1B),CCR4/MAPK信号通路(CCR4),JAK/STAT信号通路(JAK1/2/3,STAT3和STAT5B),细胞迁移(RHOA,p.N117最常见,在其他T细胞肿瘤中未见,和VAV1)和染色质重塑(ARID1A和CTCF)。虽然其中大多数对于CTCL诊断不具有特异性,但有些可能具有诊疗一体化价值,例如PLCG1或JAK/STAT基因,RHOA(p.N117I)似乎对CTCL具有特异性。
胃肠道T细胞淋巴瘤
肠道T细胞淋巴瘤(ITCL)包括两种主要亚型:肠病相关TCL(EATL)和单形性嗜上皮性肠道TCL(MEITL),以及ITCL-NOS,通过排除来诊断,没有特定的临床病理学特征。
EATL在西方人群中更为普遍,通常发生于乳糜泻患者。特征为JAK/STAT通路(JAK1和STAT3),NFkB(TNFAIP3)和表观遗传调节因子(KMT2D,BCOR和DDX3X)高频突变。
MEITL是一种罕见的ITCL,与乳糜泻无关,发生于老年患者,是亚洲主要的ITCL。STAT5B和JAK3突变最常见,且BRAF、KRAS和NRAS互斥变异的发生频率高于EATL。表观遗传基因SETD2突变也经常检测到。
肝脾T细胞淋巴瘤
肝脾T细胞淋巴瘤(HSTCL)是一种侵袭性TCL,较为罕见,发生于青少年和年轻人。HSTCL占NHL的不到1%,与慢性免疫抑制有关。通常可以检测到等臂染色体(7q),是最常见的染色体异常。7p缺失和7q扩增导致位于7号染色体上的几个癌基因(CHN2,ABCB1, PPP1R9A)表达异常。最近的研究发现了潜在的致癌驱动基因突变。SETD2、IN080和ARID1突变参与染色质修饰(与其他TCL亚型相比,几乎只发生在HSTCL中)。还检测到STAT5B、STAT3和PIK3CD突变。
乳腺植入物相关性ALCL
乳腺植入物相关性ALCL(BIA-ALCL)是一种罕见的TCL,在乳腺植入物放置较长时间(8-11年)后发生。发生于假体周围的液体和包膜。其临床、基因和分子特征与其他ALCL不同。显示克隆性TCR重排,STAT3高频突变见于高达64%的BIA-ALCL。其他突变基因包括JAK1、JAK3、DNMT3A和TP53。未在BIA-ALCA患者中发现ALK、DUSP22和TP63重排。
结外NK/T细胞淋巴瘤,鼻型
在亚洲和中南美洲,结外NK/T细胞淋巴瘤(ENKTL),鼻型,约占成熟T/NK细胞淋巴瘤的20-25%,但在欧洲和北美仅占5%。好发于上呼吸消化道。ENKTL与EBV感染有很强的相关性,尽管其作用机制尚不完全清楚。6q21-25缺失是最常见的分子异常之一,包括 PRDM1、PTPRK和FOXO3基因。JAK/STAT通路(STAT3,JAK3,STAT5B),肿瘤抑制基因(TP53,DDX3X)和表观遗传修饰因子(TET2[~5-10%],KMT2D,KMT2C)突变频率最高。
淋巴瘤液体活检
近年来,高通量测序技术已成功应用于液体活检(LB),主要是循环肿瘤DNA(ctDNA)。ctDNA是肿瘤细胞释放到体液中的游离DNA,血浆是分析最多的形式。正越来越多地应用LB,尤其是ctDNA,来支持经典诊断工具组织活检,这主要是由于该技术的优势,是一种微创方法,可以用于疾病监测,检测微小残留病灶(MRD)。在多种癌症,包括血液系统恶性肿瘤中,外周血MRD检测是有效的工具。几项研究已经证明了其用于DLBLC,cHL,MCL,PTCL的临床意义,最近的研究也证明了其用于惰性淋巴瘤(如FL)中的临床意义。
虽然突变检测的金标准是基于组织活检,但cfDNA基因检测可以作为一种补充。此外,在某些情况下,例如肿瘤无法接近或小组织活检,或者在复发、转化或其他临床事件后需要再次活检时,cfDNA检测可能是一种很好的方法。侵袭性淋巴瘤中ctDNA与FFPE检测的一致性约为80%,在肿瘤负荷低的惰性淋巴瘤中一致性略低。然而,即使在这类患者中,ctDNA分析也被证明是有效的。
然而,要使LB在临床中成功应用,从样本操作到生物信息学分析,仍需致力于使每一步都标准化。这些技术和分析因素是严峻的挑战,也是合作研究重点,从而使其最终应用于常规临床实践。
结论
随着基因组学的发展,分子数据有助于更好地诊断淋巴瘤,这些变异现在是诊断标准的一部分。NGS能够同时检测多种变异,包括突变,拷贝数变异和结构变异。基因组信息可以与形态学和免疫表型相结合,用于诊断和预后评估。然而,在淋巴瘤诊断中应用NGS的先决条件是整个过程的标准化和质量控制。
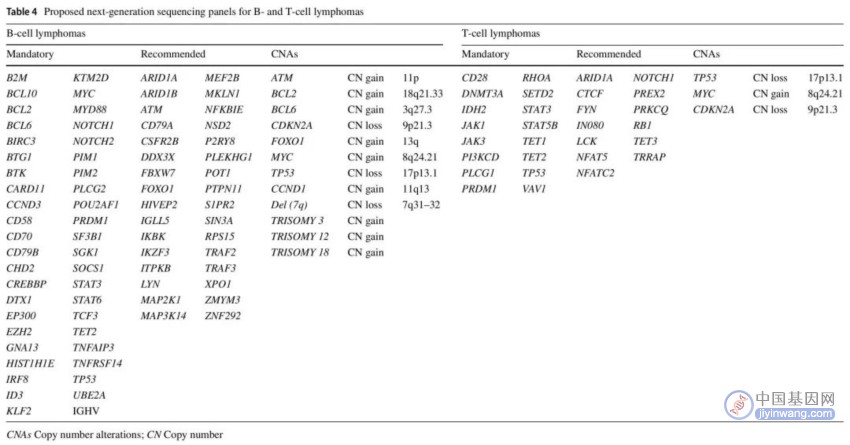
表4. B细胞和T细胞淋巴瘤NGS panel(SNV和CNA检测)
参考文献:
Sánchez-Beato M, Méndez M, Guirado M, Pedrosa L, Sequero S, Yanguas-Casás N, de la Cruz-Merino L, Gálvez L, Llanos M, García JF, Provencio M. A genetic profiling guideline to support diagnosis and clinical management of lymphomas. Clin Transl Oncol. 2023 Sep 6. doi: 10.1007/s12094-023-03307-1. Epub ahead of print. PMID: 37672206.
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。




















