Nat Commun发表人类单细胞长读长全基因组分析新方法,可为单细胞遗传变异提供新见解
在过去几年中,长读长测序技术在通量和数据质量方面取得了显著进步,极大地促进了人们对人类基因组变异的理解。长读长测序能够读取重复和高GC含量的区域,对于生成真核生物多样性的参考基因组,以及绘制完整人类基因组端粒到端粒图谱至关重要。长读长测序的另一个优点是能够对难以或无法通过其他基因组测序方法识别的复杂结构变异(SV)和重复元件进行基因分型。
人类单细胞全基因组测序(WGS)已成为一个热点研究领域,有助于解答细胞生物学多个领域的基本问题。目前,单细胞WGS研究主要集中在表征短读长测序中可检测到的遗传变异,包括单核苷酸变异(SNV)等。虽然单细胞短读长全基因组测序有多种方法,但仍未有识别单细胞全部遗传变异的技术。
近期,瑞典乌普萨拉大学等多个机构的研究人员在Nature Communications发表了题为“Long-read whole-genome analysis of human single cells”的文章,提出了一种新的人类单细胞长读长全基因组分析方法。利用基于PacBio高保真(HiFi)测序平台的长读长测序,对人类单个CD8+ T细胞进行WGS分析,并通过多重置换扩增(MDA)获得足以进行长读长测序的DNA。结果表明,与短读长测序相比,长读长测序可提高单细胞遗传变异检测的性能,特别是检测单倍型、复杂结构变异和串联重复序列(TR);并证明从头重建部分人类T细胞基因组是可行的。

文章发表在Nature Communications
兼容短读长和长读长测序的单细胞WGS工作流程
研究团队利用单细胞全基因组扩增技术(WGA)与PacBio HiFi WGS相结合开发了基于液滴的MDA技术(dMDA),对CD8+ T细胞进行分析(图1)。该单细胞WGS的具体工作流程如下,通过流式细胞荧光分选技术(FACS)分离单细胞,并将其放入含有裂解缓冲液的孔中,以便裂解细胞并释放DNA片段。随后,将DNA分子封装在约50000个液滴中,在每个液滴内发生dMDA反应。在dMDA反应过程中形成分子间嵌合体的风险大大降低,在含有单个DNA片段的液滴中,这种风险被完全消除。因此dMDA方法能够保留分子长度并减少扩增偏差。dMDA反应完成后,扩增的DNA可用于制备短读长或长读长测序文库。
在该研究中,来自同一人类供体的两个单独的CD8+T细胞(A和B)在体外进行克隆扩增,所得细胞集合用作WGA和测序的起始材料,并使用Illumina和PacBio全基因组测序分析来自克隆A和B的单细胞。
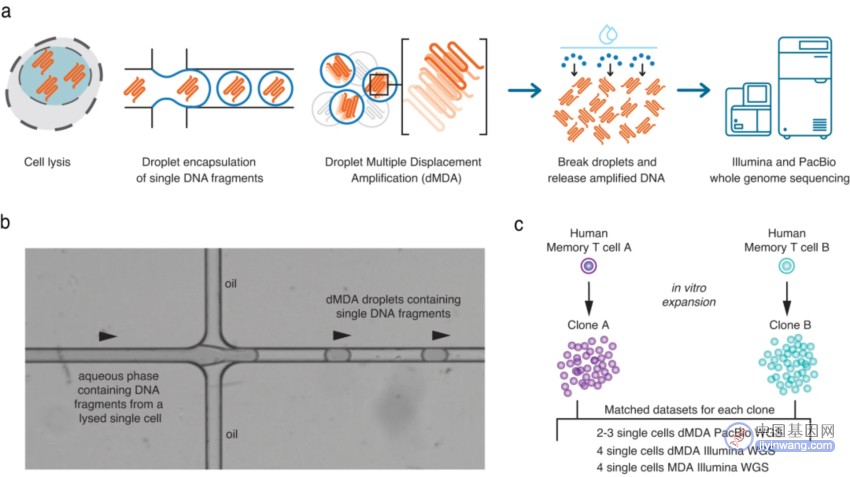
图1. 单细胞DNA扩增和测序实验概述。
dMDA增加覆盖均匀性
研究团队使用Illumina WGS分析了来自T细胞克隆A和B的16个单细胞DNA样本,其中8个样本使用dMDA进行扩增,其余8个样本则进行标准MDA处理。测序结果显示,与8个MDA样本相比,8个dMDA样本显示出更均匀的覆盖范围。在MDA样本中,68.9%的reads与≥200×覆盖率区域匹配,dMDA样本中仅为16.0%。对于完整的单细胞数据集,研究团队进一步量化了全基因组的覆盖率变化,发现MDA的标准差是dMDA样本的2.46倍。综上,与常规MDA相比,dMDA提供了更高的测序覆盖率均匀性。
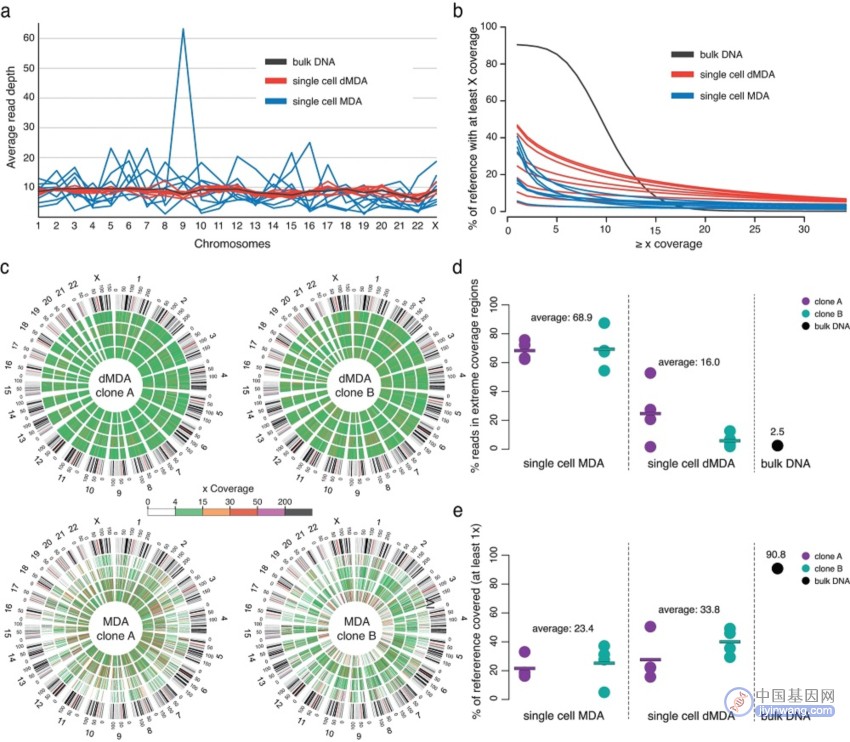
图2. MDA和dMDA用于全基因组扩增的比较。
单细胞长读长测序中SNV的检测
研究团队使用5个dMDA单细胞样本(2个来自T细胞克隆A,3个来自T细胞克隆B)进行PacBio长读长测序。结果显示,PacBio平台获得平均15.7Gb数据,Illumina平台获得48.7Gb数据。随后研究团队将该数据用于单细胞SNV调用,发现PacBio单细胞测序检测到的胚系SNV数量与Illumina相似。
进一步,研究团队估计了SNV调用的精确度和灵敏度。结果显示,PacBio单细胞SNV调用精确度高于Illumina dMDA,但灵敏度略低。在这些变异中,有6,336个位于先前报道的与人类健康相关的“暗”基因区域。值得注意的是,PacBio数据提供了数千碱基的相位信息,这使得将单细胞中检测到的变异分配给其中一种单倍型成为可能,凸显了长读长测序在单细胞水平上识别和分析体细胞变异的优势。
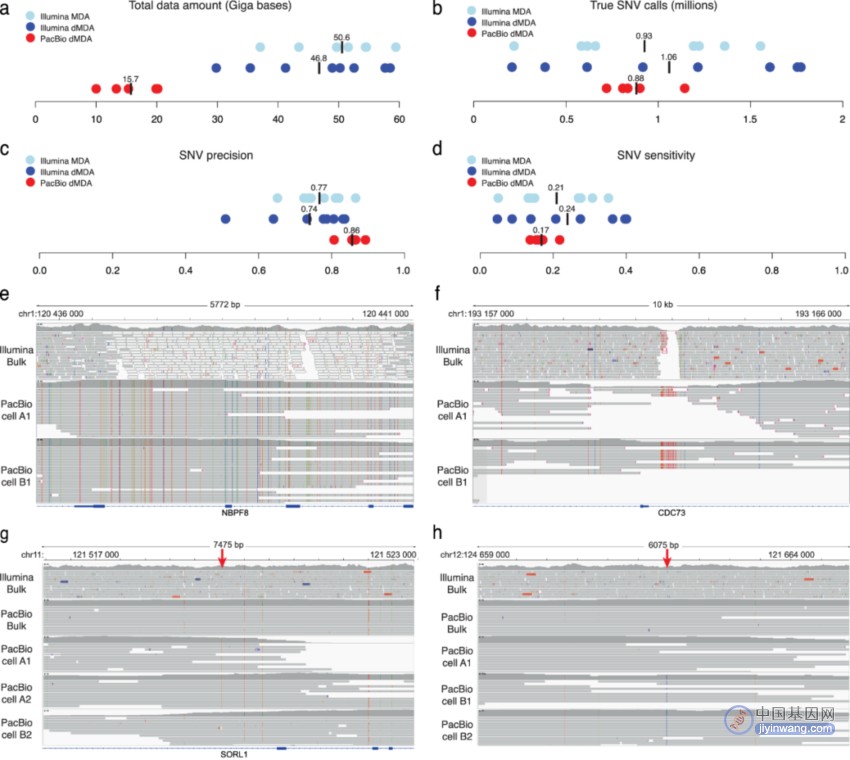
图3. 短读长和长读长测序单细胞数据中的SNV分析。
长读长单细胞WGS改进了SV的检测
研究团队使用Sniffles从PacBio数据中调用SV,发现平均每个单细胞有3,493个缺失、4,636个插入、903个重复和 19,373个倒位。由于微流控平台的缺陷,其中绝大多数SV可能源自嵌合dMDA分子。研究团队将样本间SV进行合并,以识别真正的SV,结果显示每个单细胞平均检测到5,473个真实SV,其中2510个缺失、2957个插入。
为进行比较,研究团队也对Illumina dMDA样本进行了SV分析(图4),平均检测到327个真实SV。PacBio测序检测的单细胞真实SV数量是Illumina的16倍多。PacBio检测的SV中,缺失和插入的精确度分别为0.73和0.66,其灵敏度略高于0.20;重复和倒位的精确度接近于零,表明几乎所有此类事件都源自嵌合分子。真正的PacBio SV主要由长达1kb的插入和删除组成,具有代表ALU重复元件的约300bp的清晰峰。
此外,研究团队发现在PacBio单细胞数据中检测到的一些SV在Illumina批量数据中很难被识别。PacBio数据不仅使人们能够确定这些SV在单细胞中的确切断点,还能够通过附近杂合SNV的定相来重建单倍型。
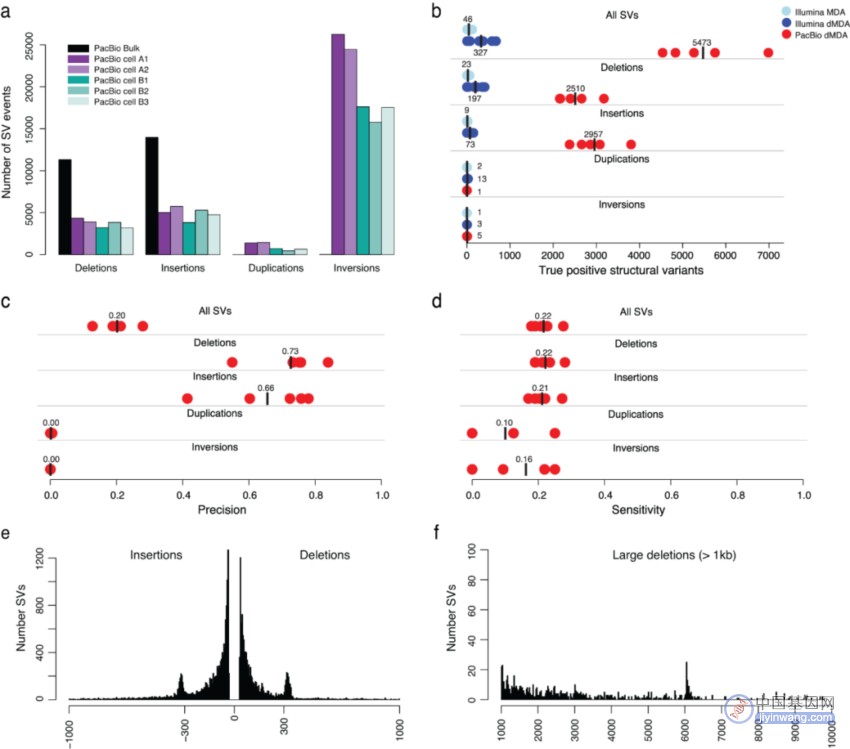
图4. 长读长单细胞数据中的SV分析。
单细胞中的TR分析
假设通过PacBio reads可研究单细胞中的TR,为证明上述观点,研究团队使用串联基因型进行TR调用(图6)。首先提取Pacbio批量数据中所有纯合或杂合的TR,共发现15,098个此类重复元件。在单细胞中,平均有4,770个TR等位基因可以被正确地进行基因分型,其重复序列大小与批量样本一致。其中,最长的重复序列比参考基因组长662bp,主要由二核苷酸AT序列组成,这是在短读长数据中难以解析的区域。
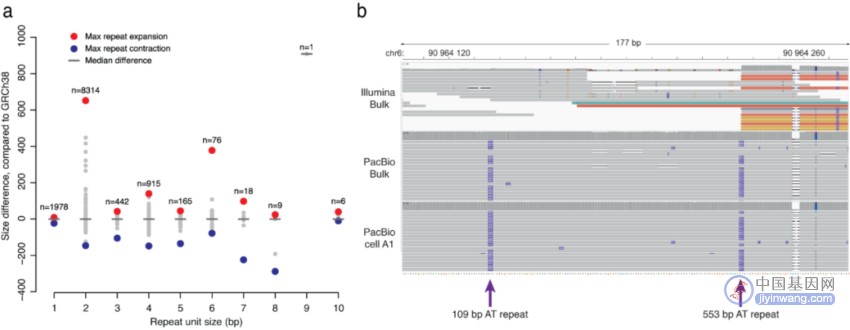
图5. 在单细胞长读数据中检测到的TR。
基于长读长数据的单细胞基因组从头组装
PacBio HiFi reads是生成高质量人类基因组组装的理想选择,为研究单细胞基因组的区域在多大程度上可以被重新构建,研究团队选择了克隆A和克隆B中覆盖率最高的两个单细胞(A1、B1),使用Hifiasm对其进行了单独的从头组装。结果显示,T细胞A1和B1分别生成598.3Mb和454.1Mb的初级片段;序列N50值分别为35kb 和42kb ,其中最大的序列N50值为578.3kb。
研究团队进一步分析了BUSCO基因模型,发现在T细胞A1中,12.8%的基因(n=1,762)被完全组装;在T细胞B1中,9.0%的基因(n=1,236)被完全组装。
结 语
综上所述,研究团队通过将自动化单细胞处理和基于液滴的WGA方法与PacBio HiFi测序相结合,能够对单个人类T细胞的长DNA片段进行测序。与短读长测序技术相比,PacBio长读长测序使每个单细胞的基因组覆盖率高达40%,从而能够检测SNV、SV和TR以及人类基因组的“暗”区域。该研究共检测到28例体细胞SNV,5,473个高置信度SV。此外,单细胞从头组装产生了高达598Mb的基因组和1762(12.8%)个完整的基因模型。总之,与短读长测序技术相比,长读长测序改进了对遗传变异的分析,包括人类基因组的“暗”区域,甚至能够从头组装部分单细胞基因组,长读长测序在表征单细胞的全谱遗传变异方面的前景。
参考文献:
Hård, J., Mold, J. E., Eisfeldt, J., Tellgren-Roth, C., Häggqvist, S., Bunikis, I., ... & Ameur, A. (2023). Long-read whole-genome analysis of human single cells.Nature Communications, 14(1), 5164.
原文链接
https://www.nature.com/articles/s41467-023-40898-3
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。












")








