科学家揭秘基因组倍增的细胞机制,为肿瘤基因组进化提供理论依据
曾京昆,成长于山东德州的一个油田小镇——临盘,从小跟着爷爷奶奶长大。
他本科就读于英国帝国理工学院生物化学系。大一和大二的暑假,曾京昆先后回国来到清华大学和北京生命科学研究所实习,大三暑假则加入葛兰素史克公司实习。

图 | 曾京昆(来源:曾京昆)
本科毕业之后,他获得英国弗朗西斯克里克研究所的全额奖学金,并来到这里攻读博士,期间师从该研究所的副所长约翰·迪弗利(John Diffley)教授,后者同时也是美国科学院院士和英国皇家学会院士。

图 | 约翰·迪弗利(John Diffley)(来源:资料图)
前不久,曾京昆刚刚发表了一篇 Cell 一作论文,这也是他在读博期间的心血之作。研究中,他和同事建立了可以过度表达 Cyclin E 的细胞模型,借此揭示了连接复制压力和全基因组加倍的机制。
01
完善学界对于癌症全基因组加倍机制的理解
据了解,90% 的人类癌症都存在染色体数目异常的现象。在一些癌症中,有丝分裂中个别染色体的错配,会导致少量染色体的增加或减少。
然而,在许多其他癌症中,染色体数目要高得多,并且它们的核型常被描述为亚四倍体,这种广泛的非整倍体很可能是从四倍体中间体生成的。
对于从二倍体变为四倍体这种染色体数目加倍现象,人们称之为全基因组加倍(whole genome duplication,WGD)。历史上,大约 30% 至 40% 的人类肿瘤经历了全基因组加倍 [1],这也是肿瘤发生过程中最常见的基因组事件。
全基因组加倍与不良预后有着较大关系,患有全基因组加倍肿瘤的病人,他们的生存预期往往更短。因此,了解全基因组加倍的原因和后果,对于癌症生物学来说十分重要。
然而,对于全基因组加倍的分子机制和细胞机制,人们了解得还够不清晰。
之前,有癌症基因组学研究表明 [1-2],Rb-E2F 通路的失调(比如 cyclin E 基因的扩增),与肿瘤的全基因组加倍有关。但是,其背后的分子机制和细胞机制依旧尚不明确。
Cyclin E 是一个调节细胞周期转换的蛋白质。在许多肿瘤中都曾出现过 Cyclin E 基因的扩增。
目前,已知可以造成全基因组加倍的方式大概有三种:病毒感染导致的细胞融合、胞质分裂失败、以及有丝分裂绕过后的内源性复制。
通过使用荧光活细胞延时摄影技术,曾京昆发现过度表达 Cyclin E 的细胞,会通过绕过有丝分裂来进行全基因组加倍。这种绕过有丝分裂的方式,是由于复制压力引起的。即在 Cyclin E 的过度表达里,扰乱复制起始位点的使用会引起复制压力。
该团队发现在其他形式的复制压力下,比如抑制 DNA 聚合酶或反应性氧化物中,细胞也会绕过有丝分裂。而在其他形式的复制压力下,细胞绕过有丝分裂之后,会进入细胞衰老仅而会停止生长。
然后,Cyclin E 会进一步克服这种细胞衰老,让细胞进行完整的内源性复制,仅而达到全基因组加倍。
在由曾京昆担任主力的这项研究里,他和同事的一个重要发现是:复制压力诱导的全基因组加倍取决于 p53 的存在。
p53 是癌症中最常发生突变的基因,也是公认的保卫基因组完整性的抑癌基因。然而,该课题组发现在全基因组加倍这样一种破坏基因组的完整性事件中,p53 竟然发挥了促进作用。
他们发现在复制压力下,p53 通过上调 CDK 抑制剂 p21,催生 APC/CCdh1 的激活,进而降解 G2 标记物,以此来促进有丝分裂的绕过。
此前曾被广泛研究的肿瘤抑制基因 p53,竟然在全基因组加倍中发挥促进作用,这一发现非常令人意外,但也能促使人们重新认识 p53 在癌症进化中的作用。
了解全基因组加倍的机制,既有助于理解癌症的产生,也有助于评估癌症药治疗的效果。因此,该成果不仅完善了学界对于癌症全基因组加倍机制的理解,也为癌症进化带来了新思考。
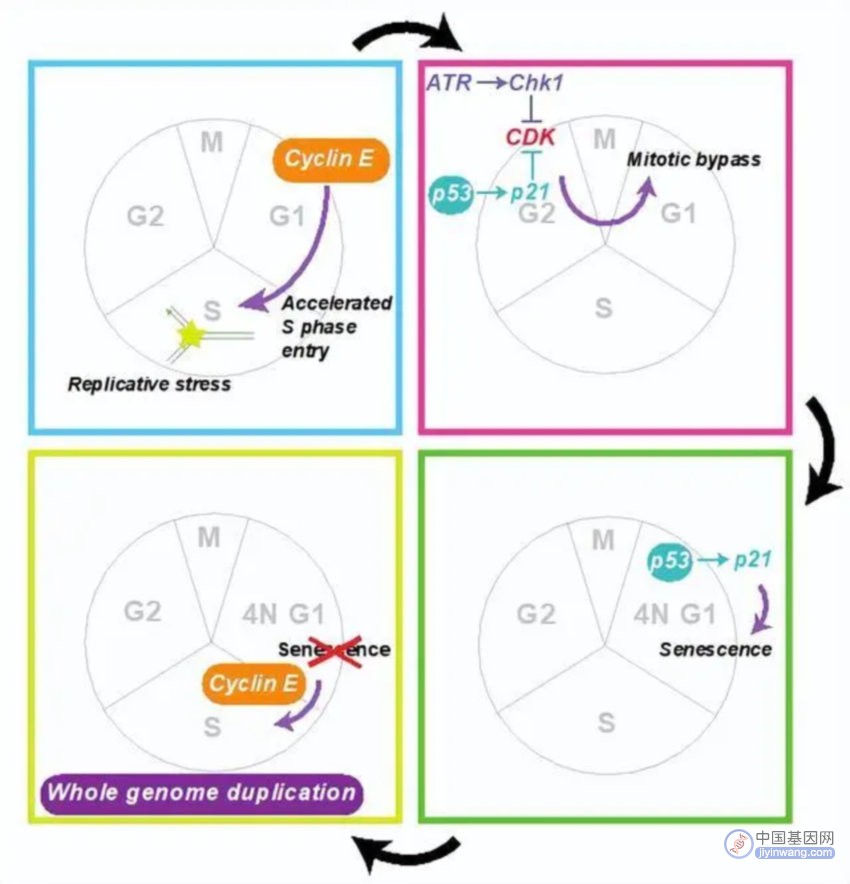
图 | 本次研究的梗概图(来源:Cell)
尽管这是一个基础研究项目,但也能为癌症治疗带来新思路。原因在于,常见化疗药物是通过阻碍 DNA 复制、制造复制压力以杀死癌细胞,所以化疗药物也许会导致肿瘤全基因组加倍的产生。
而基于本次工作,人们或许可以找到多倍体细胞的弱点,通过靶向性地杀死多倍体细胞的方法,实现恶性肿瘤的治疗。
另据悉,CDK4/6 抑制剂是一种新型靶向治疗药物,其有效性已在多种癌症治疗中得到证明。而 CDK4/6 是 Rb-E2F 中的一环,主要通过和另一个细胞周期蛋白 cyclin D 的结合,来促进细胞周期的进展。
本次研究亦表明 cyclin E 的过表达、以及其他一些 Rb-E2F 通路中因子失调所存在的危害性。而 cyclin E 会和 CDK2 结合,CDK2 的激活正是 CDK4/6 一个常见的耐药机制。
这样来看,CDK2 抑制剂与化疗手段的结合,有望防止全基因组加倍的产生,进而提高癌症治疗效果。或者,也可以通过 cyclin E 和 CDK4/6 抑制剂的结合,来增强疗效以及防止耐药性。

图 | 相关论文(来源:Cell)
日前,相关论文以《细胞周期蛋白 E 诱导的复制应激驱动 p53 依赖性全基因组复制》(Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication)为题发在,曾京昆是第一作者,约翰·迪弗利(John Diffley)担任通讯作者 [3]。
02
“这不正是我们需要的吗?”
曾京昆表示,此次研究出于偶然,甚至有些歪打正着。在他博士入学时,当时在该团队做博后研究的斯蒂芬妮·黑欧斯(Stephanie Hills)正在研究 cyclin E 对于 DNA 复制起始的影响。
后来,斯蒂芬妮发现一部分 cyclin E 过表达的细胞变成了四倍体,但却始终找不到原因。
而曾京昆对这个现象很感兴趣,在斯蒂芬妮离职之后便把它作为备胎项目来研究。
“阴差阳错的是,我的主项目后来被人抢发了,结果我这个备胎项目反而越来越有眉目,当然也跟我们实验室的方向越来越偏离。”曾京昆说。
总而言之,一开始课题组并没有刻意设计课题,也不知道全基因组加倍是什么,纯粹是带着兴趣出发。
曾京昆表示:“尽管我们组算是全球最好的生化实验室之一,但是在细胞生物学上的研究并不多,也没有什么细胞实验的手段,所以一切都是从零摸索。”
如前所述,目前已知有三种方式可以造成全基因组加倍。因此,曾京昆的第一件事是搞清楚 cyclin E 过表达,到底是通过哪种方式造成了全基因组加倍。
后来,他在一个学术会议上看到了基于 FUCCI(fluorescent ubiquitination–based cell-cycle indicator)的活细胞延时摄影技术,它能对活细胞中的不同细胞周期阶段进行荧光标记。
“我心里就想这不正是我们需要的吗?于是就把 FUCCI 技术引入实验室,并改进得让它可以区分有丝分裂期。后来,我们通过这项技术发现,cyclin E 过表达的细胞是通过有丝分裂绕过,实现了全基因组加倍。”曾京昆说。
项目中的一个意外发现是,有丝分裂绕过竟然依赖于 p53。p53 是一个维护基因组稳定性的抑癌基因。而抑制全基因组加倍,会损伤基因组的完整性。
因此,曾京昆一开始想的是,敲除掉 p53 应该更有利于全基因组加倍。结果他却发现在 cyclin E 过表达之下,当敲除 p53 之后,全基因组加倍反而减少。
使用 FUCCI 技术观测之后发现,细胞丧失了有丝分裂绕过的能力,并在进入灾难性有丝分裂之后死亡。这一意外发现让他得出了如下结论:p53 可以促进有丝分裂绕过。
这项研究贯穿了曾京昆几乎整个博士生涯。博三开学时,有一次大家和导师约翰一起闲聊喝酒。微醺之中,约翰开始点评大家的项目进展,对于前面几位同事的项目,约翰一一给出了自己的见解。
轮到点评曾京昆的项目时,约翰开玩笑地说:“I have no idea about what you’re doing。”
回忆此事曾京昆表示:“其实他说的也是事实,一来我的项目跟实验室的主题毫不相干,二来我那时的实验结果很零散也很难解读。听到他的玩笑之后我也笑了,但并没有感到气馁,不过也确实让我意识到应该好好梳理一下思路。”于是,通过一番认真取舍,曾京昆把实验结果串成一条故事线,最终获得了导师的认可。
他继续说道:“读博期间,我还得到了 BIF fellowship,这是欧洲很有竞争力的一个 fellowship,只有不到 10% 的申请者能够获得。目前我的博四已经结束,正在准备论文答辩。”
接下来,除了继续推进基础研究之外,他打算开发一些靶向治疗方法,比如靶向多倍体细胞以及靶向基因突变。目前,曾京昆已经酝酿出来一些想法。在博士毕业之后,他将寻找合适的实验室做博后研究。“等到学有所成之后,我会考虑回国。国内有不少高水平的科研院所,它们都在我的考虑范围之内。”曾京昆最后表示。
参考资料:
1.Bielski, C.M. et al. (2018). Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 50, 1189–1195.
2.Zack, T.I. et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140.
3.Zeng, J., Hills, S. A., Ozono, E., & Diffley, J. F. (2023). Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication. Cell.
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。












")








