日本人真是“徐福”后代?Science子刊发布基因组研究,发现日本人的三种祖先起源!

随着全基因组测序(WGS)技术的发展,人类遗传学和生物医学研究获得了前所未有的深入洞察。WGS数据集不仅揭示了人类基因组变异的特性,还阐明了人类种群的复杂历史,并为进化适应和正向选择的过程提供了线索。
近日,一个由日本遗传学家、基因组学和基因分型专家组成的多机构团队对来自日本全国各地的数千名日本人的基因组进行了测序,创建了日本全基因组/外显子测序库(JEWEL),旨在深入理解日本人群的遗传特征。
相关研究论文以Decoding triancestral origins, archaic introgression, and natural selection in the Japanese population by whole- genome sequencing为题发表在Science Advances杂志。

先说结论
(1)该研究是迄今为止规模最大的非欧洲人测序研究之一。研究人员对居住在日本和冲绳七个地区的 3,200 多人进行了基因组测序,创建了新的日本全基因组/外显子组测序库 (JEWEL)。
(2)研究发现,现代日本人的祖先有三个主要族群:新石器时代的绳文狩猎采集者、汉族人的祖先,以及一个未知的东北亚人群。
(3)研究还发现,现代日本人的基因中有来自丹尼索瓦人和尼安德特人的基因,其中一些基因与某些疾病的发展有关,如二型糖尿病、前列腺癌、冠心病和类风湿性关节炎。识别这些变异基因可以为日本人提供更好的医疗保健。
JEWEL数据集的特点
JEWEL数据集包含了来自日本七个地理区域的3256个个体的高深度全基因组测序数据。通过标准Illumina协议进行测序,平均基因组覆盖度达到25.6倍。数据集最终包含45,586,919个单核苷酸变异(SNVs)和9,113,420个插入或缺失(indels)。
与基因组聚合数据库(gnomAD)和ToMMo项目相比,JEWEL中有61%和40%的变异是未被记录的,其中15,410,953(32.7%)变异是JEWEL独有的。这些结果确认了JEWEL数据集在多个方面的高质量,为深入分析日本人群的遗传特性提供了基础。
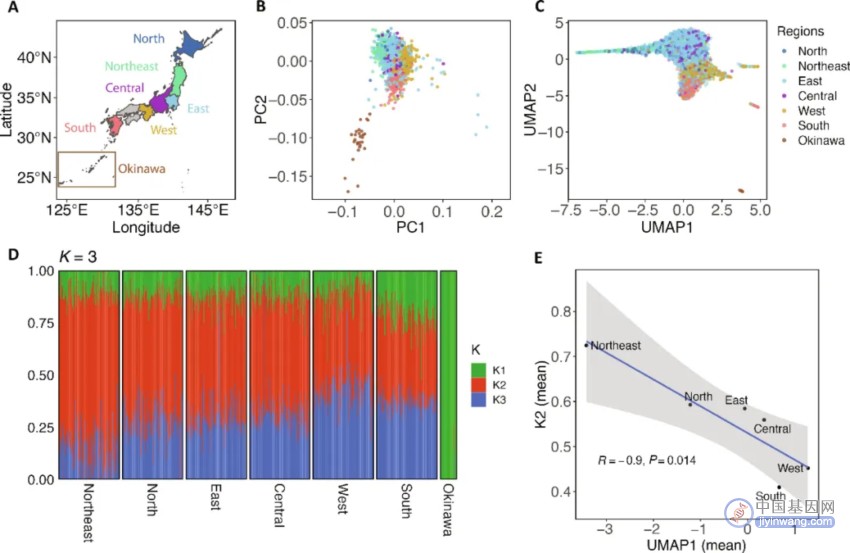
日本人群的三源起源
通过主成分分析(PCA)和基于罕见变异的PCA-UMAP分析,研究揭示了日本人群的精细遗传结构。这些分析不仅重现了基于常见变异的PCA分析的经典“双聚类”结构,还突出了几个显著特征,包括本州岛各亚区的更清晰分离、琉球群岛与本州岛的更明显区分,以及西部和南部个体的额外亚聚类。
ADMIXTURE分析表明,当代日本人群可以最佳地被建模为三个祖源成分(K1至K3)的混合。通过与现代和古代遗传数据的比较,研究估计了Jomon、东亚(EA)和东北亚(NEA)祖先在日本各亚群中的贡献,为理解日本人群的三源起源提供了证据。
功能丧失(LoF)变异和人类敲除
JEWEL数据集允许研究者探索在日本人群中可能具有临床重要性的蛋白质编码变异。研究中鉴定了18,481个LoF变异,涉及9045个基因,其中包括未在gnomAD或ToMMo中登记的9780个LoF变异。
此外,研究还鉴定了可能具有临床相关性的人类敲除(homozygotes或compound heterozygotes for LoF variants)。
尼安德特人和丹尼索瓦人基因渗入序列
研究应用了一种新开发的计算方法IBDMix,以检测可能来自尼安德特人或丹尼索瓦人的渗入序列。研究在日本人群中识别了与49个表型相关的44个古老片段,其中大多数片段在之前的研究中未曾报道。
特别是,与尼安德特人衍生的NKX6-1片段相关的T2D,以及与丹尼索瓦人衍生的POLR3E片段相关的身高,为理解古老基因渗入对现代人类表型影响提供了新见解。
研究意义
在日本人群的研究中,由于以往数据的欧洲中心性,对于东亚人群,特别是日本人群的遗传多样性了解仍然有限。
所以,不管日本人祖先是不是真的是“徐福”,这项研究对于理解日本人群的遗传结构、疾病风险以及对古老基因渗入的适应都具有重要意义。同时,它也为精准医疗提供了丰富的遗传资源,有助于开发针对日本人群的个性化医疗策略。
此外,通过比较不同地区和不同人群的遗传数据,研究还揭示了人类进化过程中的自然选择和适应性变化,为人类学和进化生物学的研究提供了新的视角和数据支持。
参考文献:https://www.science.org/doi/10.1126/sciadv.adi8419
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。




















